
Contrasting Epidemiology and Population Genetics of COVID-19 Infections Defined by Multilocus Genotypes in SARS-CoV-2 Genomes Sampled Globally


Abstract
:1. Introduction
2. Material and Methods
2.1. Data Curation
2.2. Study Variables
2.3. Sequence Alignments and Multilocus Genotyping
2.4. Genetic Diversity Indices
2.5. Linkage Disequilibrium Estimates
2.6. Genetic Differentiation Estimates
2.7. Statistical Analysis
3. Results and Discussion
3.1. Demographics of the Study Population
3.2. Polymorphic Loci Investigated
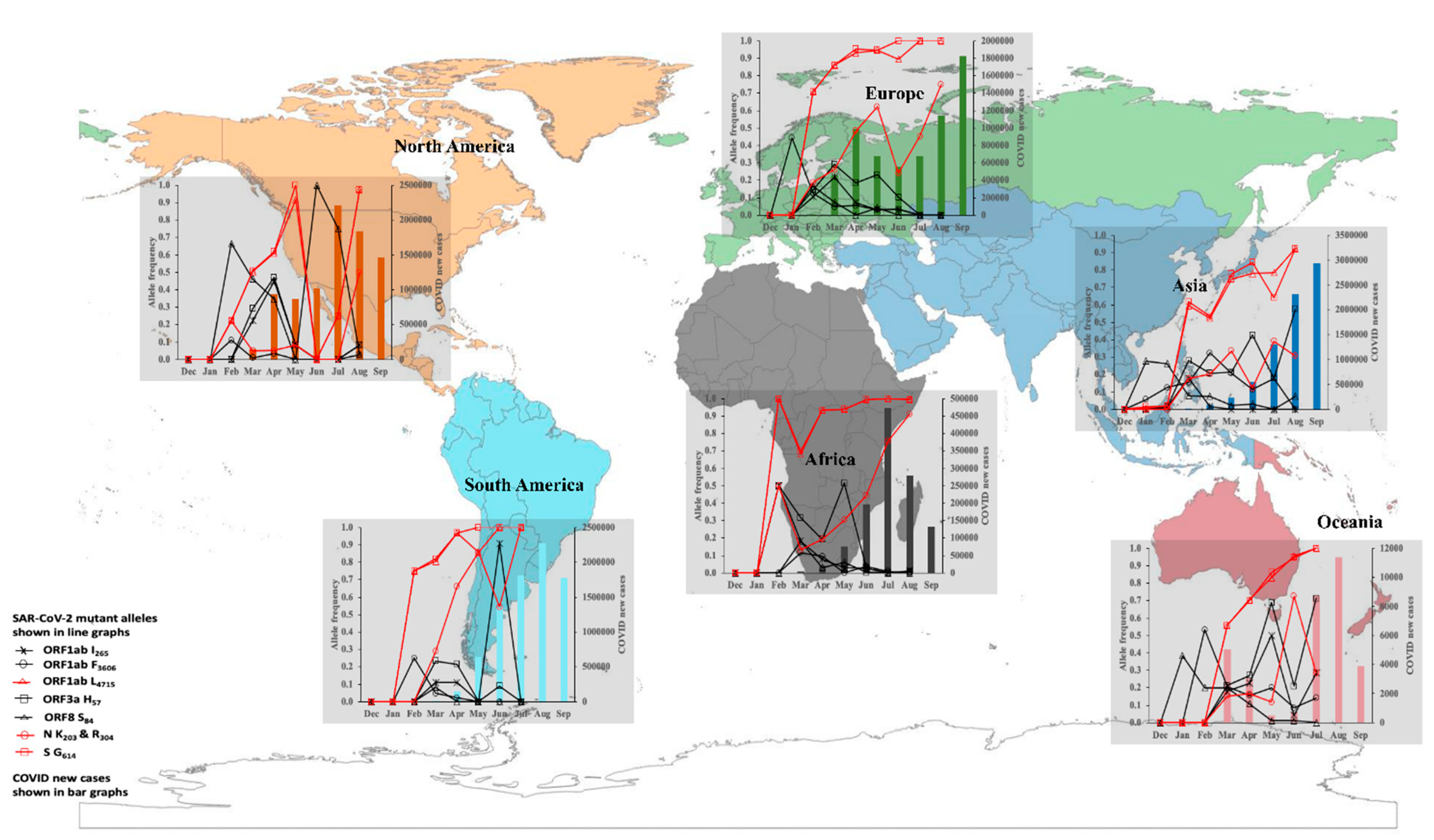
4. SARS-CoV-2 Transmission Dynamics and Epidemiological Risk of Infection in 2020
5. Population Genetics of SARS-CoV-2 Infections in 2020
6. Genetic Clustering and Differentiation of SARS-CoV-2 Populations in 2020 and 2021
7. Conclusions
8. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Velavan, T.P.; Pallerla, S.R.; Rüter, J.; Augustin, Y.; Kremsner, P.G.; Krishna, S.; Meyer, C.G. Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine 2021, 72, 103629. [Google Scholar] [CrossRef]
- Suh, S.; Lee, S.; Gym, H.; Yoon, S.; Park, S.; Cha, J.; Kwon, D.-H.; Yang, Y.; Jee, S.H. A systematic review on papers that study on Single Nucleotide Polymorphism that affects coronavirus 2019 severity. BMC Infect. Dis. 2022, 22, 47. [Google Scholar] [CrossRef]
- Talic, S.; Shah, S.; Wild, H.; Gasevic, D.; Maharaj, A.; Ademi, Z.; Li, X.; Xu, W.; Mesa-Eguiagaray, I.; Rostron, J.; et al. Effectiveness of public health measures in reducing the incidence of covid-19, SARS-CoV-2 transmission, and covid-19 mortality: Systematic review and meta-analysis. BMJ 2021, 375, e068302. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Liu, M.; Wu, Z.; Liu, Y.; Li, W.; Liu, M.; Wang, X.; Gao, B.; Luo, Y.; et al. The Effect of the COVID-19 Vaccine on Daily Cases and Deaths Based on Global Vaccine Data. Vaccines 2021, 9, 1328. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chi, W.-J.; Lin, Y.-T.; Lai, C.-Y. The spatiotemporal estimation of the risk and the international transmission of COVID-19: A global perspective. Sci. Rep. 2020, 10, 20021. [Google Scholar] [CrossRef]
- ECDC. SARS-CoV-2 Variants of Concern as of 11 March 2022. 2022. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 18 March 2022).
- Zhao, Y.; Huang, J.; Zhang, L.; Chen, S.; Gao, J.; Jiao, H. The global transmission of new coronavirus variants. Environ. Res. 2022, 206, 112240. [Google Scholar] [CrossRef]
- McArthur, L.; Sakthivel, D.; Ataide, R.; Chan, F.; Richards, J.S.; Narh, C.A. Review of Burden, Clinical Definitions, and Management of COVID-19 Cases. Am. J. Trop. Med. Hyg. 2020, 103, 625–638. [Google Scholar] [CrossRef]
- Dimeglio, C.; Nicot, F.; Miedougé, M.; Chappert, J.-L.; Donnadieu, C.; Izopet, J. Influence of age on the spread of SARS-CoV-2 variant B.1.1.7. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2021, 141, 104872. [Google Scholar] [CrossRef]
- Harper, H.; Burridge, A.; Winfield, M.; Finn, A.; Davidson, A.; Matthews, D.; Hutchings, S.; Vipond, B.; Jain, N.; The COVID-19 Genomics UK (COG-UK) Consortium; et al. Detecting SARS-CoV-2 variants with SNP genotyping. PLoS ONE 2021, 16, e0243185. [Google Scholar] [CrossRef]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, M.; Chen, K.; Cheng, L.; Weng, T.; Shi, D.; et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 2020, 6, 76. [Google Scholar] [CrossRef]
- Banoun, H. Evolution of SARS-CoV-2: Review of Mutations, Role of the Host Immune System. Nephron 2021, 145, 392–403. [Google Scholar] [CrossRef]
- Bhat, S.; Pandey, A.; Kanakan, A.; Maurya, R.; Vasudevan, J.S.; Devi, P.; Chattopadhyay, P.; Sharma, S.; Khyalappa, R.J.; Joshi, M.G.; et al. Learning From Biological and Computational Machines: Importance of SARS-CoV-2 Genomic Surveillance, Mutations and Risk Stratification. Front. Cell. Infect. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Pang, X.; Li, P.; Zhang, L.; Que, L.; Dong, M.; Xie, B.; Wang, Q.; Wei, Y.; Xie, X.; Li, L.; et al. Emerging Severe Acute Respiratory Syndrome Coronavirus 2 Mutation Hotspots Associated With Clinical Outcomes and Transmission. Front. Microbiol. 2021, 12, 753823. [Google Scholar] [CrossRef]
- Morais, I.J.; Polveiro, R.C.; Souza, G.M.; Bortolin, D.I.; Sassaki, F.T.; Lima, A.T.M. The global population of SARS-CoV-2 is composed of six major subtypes. Sci. Rep. 2020, 10, 18289. [Google Scholar] [CrossRef]
- Justo Arevalo, S.; Zapata Sifuentes, D.; Huallpa, C.J.; Landa Bianchi, G.; Castillo Chávez, A.; Garavito-Salini Casas, R.; Uceda-Campos, G.; Pineda Chavarria, R. Global Geographic and Temporal Analysis of SARS-CoV-2 Haplotypes Normalized by COVID-19 Cases During the Pandemic. Front. Microbiol. 2021, 12, 612432. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- ISARIC. COVID-19 Report 2020. International Severe Acute Respiratory and Emerging Infections Consortium. Available online: https://media.tghn.org/medialibrary/2020/05/ISARIC_Data_Platform_COVID-19_Report_27APR20.pdf (accessed on 20 January 2022).
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2014, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P. vegan: Community Ecology Package; R package version 2.4-3; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Mangin, B.; Siberchicot, A.; Nicolas, S.; Doligez, A.; This, P.; Cierco-Ayrolles, C. Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity 2012, 108, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.; Glassberg, E.C.; Harpak, A.; Feldman, M.W. Clonal interference can cause wavelet-like oscillations of multilocus linkage disequilibrium. J. R. Soc. Interface 2018, 15, 20170921. [Google Scholar] [CrossRef]
- Winter, D. MMOD: An R library for the calculation of population differentiation statistics. Mol. Ecol. Resour. 2012, 12, 1158–1160. [Google Scholar] [CrossRef]
- Hedrick, P.W. A Standardized Genetic Differentiation Measure. Evolution 2005, 59, 1633–1638. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant graphics for data analysis. J. Stat. Softw. 2010, 35, 65–88. [Google Scholar]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef] [Green Version]
- RCore. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org/ (accessed on 18 December 2021).
- StataCorp. Stata Statistical Software. 2019. Available online: https://www.stata.com/company/ (accessed on 15 January 2021).
- Leung, D.T.; Tam, F.C.; Ma, C.H.; Chan, P.K.; Cheung, J.L.; Niu, H.; Tam, J.S.; Lim, P.L. Antibody response of patients with severe acute respiratory syndrome (SARS) targets the viral nucleocapsid. J. Infect. Dis. 2004, 190, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Swanson, S.E.; Negatu, S.G.; Dittmar, M.; Miller, J.; Ramage, H.R.; Cherry, S.; Jurado, K.A. SARS-CoV-2 viral proteins NSP1 and NSP13 inhibit interferon activation through distinct mechanisms. PLoS ONE 2021, 16, e0253089. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Ramesh, S.; Govindarajulu, M.; Parise, R.S.; Neel, L.; Shankar, T.; Patel, S.; Lowery, P.; Smith, F.; Dhanasekaran, M.; Moore, T. Emerging SARS-CoV-2 Variants: A Review of Its Mutations, Its Implications and Vaccine Efficacy. Vaccines 2021, 9, 1195. [Google Scholar] [CrossRef]
- Russell, T.W.; Wu, J.T.; Clifford, S.; Edmunds, W.J.; Kucharski, A.J.; Jit, M. Effect of internationally imported cases on internal spread of COVID-19: A mathematical modelling study. Lancet Public Health 2021, 6, e12–e20. [Google Scholar] [CrossRef]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill. Bull. Eur. Sur Les Mal. Transm. Eur. Commun. Dis. Bull. 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Trauer, J.M.; Lydeamore, M.J.; Dalton, G.W.; Pilcher, D.; Meehan, M.T.; McBryde, E.S.; Cheng, A.C.; Sutton, B.; Ragonnet, R. Understanding how Victoria, Australia gained control of its second COVID-19 wave. Nat. Commun. 2021, 12, 6266. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef]
- Poterico, J.A.; Mestanza, O. Genetic variants and source of introduction of SARS-CoV-2 in South America. J. Med. Virol. 2020, 92, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sokhansanj, B.A.; Malhotra, C.; Zheng, K.; Rosen, G.L. Genetic grouping of SARS-CoV-2 coronavirus sequences using informative subtype markers for pandemic spread visualization. PLOS Comput. Biol. 2020, 16, e1008269. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-L.; Dichio, V.; Rodríguez Horta, E.; Thorell, K.; Aurell, E. Global analysis of more than 50,000 SARS-CoV-2 genomes reveals epistasis between eight viral genes. Proc. Natl. Acad. Sci. USA 2020, 117, 31519–31526. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.; et al. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef]
- Rockett, R.J.; Arnott, A.; Lam, C.; Sadsad, R.; Timms, V.; Gray, K.-A.; Eden, J.-S.; Chang, S.; Gall, M.; Draper, J.; et al. Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat. Med. 2020, 26, 1398–1404. [Google Scholar] [CrossRef]
- Sun, H.; Dickens, B.L.; Cook, A.R.; Clapham, H.E. Importations of COVID-19 into African countries and risk of onward spread. BMC Infect. Dis. 2020, 20, 598. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef]
- Seemann, T.; Lane, C.R.; Sherry, N.L.; Duchene, S.; Gonçalves da Silva, A.; Caly, L.; Sait, M.; Ballard, S.A.; Horan, K.; Schultz, M.B.; et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat. Commun. 2020, 11, 4376. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- GISAID. Clade and Lineage Nomenclature Aids in Genomic Epidemiology Studies of Active hCoV-19 Viruses. 2022. Available online: https://www.gisaid.org/resources/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/ (accessed on 19 April 2022).
- Schmidt, M.; Arshad, M.; Bernhart, S.H.; Hakobyan, S.; Arakelyan, A.; Loeffler-Wirth, H.; Binder, H. The Evolving Faces of the SARS-CoV-2 Genome. Viruses 2021, 13, 1764. [Google Scholar] [CrossRef]
- Thakur, V.; Bhola, S.; Thakur, P.; Patel, S.K.S.; Kulshrestha, S.; Ratho, R.K.; Kumar, P. Waves and variants of SARS-CoV-2: Understanding the causes and effect of the COVID-19 catastrophe. Infection 2022, 50, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Global | Africa | Asia | Europe | North America | Oceania | South America | ||
|---|---|---|---|---|---|---|---|---|
| Characteristics | 5959 | 601 | 1579 | 1188 | 597 | 1646 | 348 | |
| Age group # (years) | 0–19 | 323 (5.4) | 68 (11.3) | 123 (7.8) | 43 (3.6) | 20 (3.6) | 58 (3.5) | 11 (3.2) |
| 20–39 | 2054 (34.5) | 276 (45.9) | 559 (35.4) | 273 (22.9) | 196 (32.8) | 634 (38.5) | 116 (33.3) | |
| 40–59 | 1996 (33.5) | 185 (30.8) | 561 (35.5) | 389 (32.7) | 217 (36.4) | 515 (31.3) | 129 (37.1) | |
| 60+ | 1586 (26.6) | 72 (11.9) | 336 (21.3) | 483 (40.7) | 164 (27.5) | 439 (26.7) | 92 (26.4) | |
| Gender # | Females | 2664 (44.7) | 338 (56.2) | 581 (36.8) | 581 (48.9) | 250 (41.9) | 754 (45.8) | 160 (45.9) |
| Males | 3295 (55.3) | 263 (43.8) | 998 (63.2) | 607 (51.1) | 347 (58.1) | 892 (54.2) | 188 (54.0) | |
| Clinical # Severity | Asymptomatic | 60 (1.5) | 0 (0.0) | 41 (2.7) | 18 (1.9) | 0 (0.0) | 0 (0.0) | 1 (0.4) |
| Mild | 557 (14.1) | 15 (2.5) | 134 (8.8) | 64 (6.7) | 282 (47.2) | 2 (12.5) | 60 (24.9) | |
| Severe | 3321 (84.33) | 586 (97.5) | 1356(88.6) | 870 (91.4) | 315 (52.8) | 14 (87.5) | 180 (74.7) | |
| Missing data * | 2021 | 0 | 48 | 236 | 0 | 1630 | 107 | |
| Continent | N | MLGs | eMLGs (SE) | E.5 | He | d (p-Value) | d-cc (p-Value) |
|---|---|---|---|---|---|---|---|
| Africa | 601 | 74 | 56.4 (3.1) | 0.57 | 0.19 | 0.037 (0.001) | 0.009 (0.180) |
| Asia | 1579 | 185 | 78.1(5.2) | 0.48 | 0.26 | 0.084 (0.001) | 0.024 (0.001) |
| Europe | 1188 | 97 | 53.2 (3.8) | 0.41 | 0.18 | 0.088 (0.001) | 0.011 (0.107) |
| North America | 597 | 69 | 48.6 (3.3) | 0.47 | 0.30 | 0.234 (0.001) | 0.073 (0.001) |
| Oceania | 1646 | 181 | 77.5 (5.0) | 0.45 | 0.30 | 0.087 (0.001) | 0.023 (0.001) |
| South America | 348 | 40 | 40.0 (0.0) | 0.60 | 0.15 | 0.091 (0.001) | 0.014 (0.132) |
| Total | 5959 | 472 | 95.3 (5.8) | 0.34 | 0.26 | 0.086 (0.001) | 0.023 (0.001) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, F.H.M.; Ataide, R.; Richards, J.S.; Narh, C.A. Contrasting Epidemiology and Population Genetics of COVID-19 Infections Defined by Multilocus Genotypes in SARS-CoV-2 Genomes Sampled Globally. Viruses 2022, 14, 1434. https://doi.org/10.3390/v14071434
Chan FHM, Ataide R, Richards JS, Narh CA. Contrasting Epidemiology and Population Genetics of COVID-19 Infections Defined by Multilocus Genotypes in SARS-CoV-2 Genomes Sampled Globally. Viruses. 2022; 14(7):1434. https://doi.org/10.3390/v14071434
Chicago/Turabian StyleChan, Felicia Hui Min, Ricardo Ataide, Jack S. Richards, and Charles A. Narh. 2022. "Contrasting Epidemiology and Population Genetics of COVID-19 Infections Defined by Multilocus Genotypes in SARS-CoV-2 Genomes Sampled Globally" Viruses 14, no. 7: 1434. https://doi.org/10.3390/v14071434