1. Introduction
Vegetables are a major dietary source of many essential nutrients and fibre in a balanced human diet. The Australian vegetable industry is a significant part of the national economy, providing both fresh and processed vegetables and vegetable products for domestic consumption and for export. With a gross value of
$4.1 billion, vegetable production was Australia’s sixth highest-value agricultural industry, with exports accounting for
$385 million of the country’s agricultural export revenue in 2017–18 [
1]. In Tasmania, the vegetable sector contributes substantially to the state’s economy, with a gross value of
$299 million in 2019–20 and with up to
$21 million of export revenue [
2]. However, numerous plant pathogens, including viruses, can infect vegetables, posing a significant threat to crop growth, yield, quality, and market value.
Green peas are grown on significant areas of Tasmanian land and are known to host a variety of viruses from various families, including
Luteoviridae,
Solemoviridae,
Potyviridae and others (
Table 1) [
3,
4]. Chlorosis, vein clearing, mottling, dwarfing, enations, and necrosis are some of the symptoms caused by these viruses in single or mixed infections [
5,
6,
7,
8,
9]. Among the viruses most commonly found infecting green pea in Tasmania are members of the
Polerovirus genus, in particular
Turnip yellows virus (TuYV) [
3]. The
Polerovirus genus (family
Solemoviridae) is genetically diverse, with 32 approved or tentative species [
10] that share biological characteristics such as persistent transmission by insect vectors (primarily aphids) and phloem-limitation in the plant hosts [
11,
12,
13]. Examination of virus abundance, distribution, diversity, and alternative hosts will be critical for developing appropriate management strategies [
3,
14].
TuYV was first reported in the United Kingdom as a European strain of
Beet western yellows virus (BWYV) based on biology and serology [
15]. TuYV and BWYV were later classified as separate species by the International Committee on Taxonomy of Viruses (ICTV) based on different host ranges [
16]. TuYV has a monopartite linear single-stranded RNA (ssRNA) genome that contains seven overlapping open reading frames (ORF0 to ORF5 and ORF3a). These ORFs encode proteins associated with different functions, including suppression of the host silencing response and virus accumulation (P0), virus replication (P1–P2), systemic infection and phloem-limitation (P3a and P4) and capsid proteins and vector recognition (P3 and P3–P5) [
17,
18,
19,
20,
21,
22,
23]. P3 is the major coat protein (CP) essential for RNA stability and virion assembly. The P3–P5 fusion of CP with the P5 readthrough domain (RTD) is a minor component of the capsid. The RTD, exposed on the virion surface, is not required for infection initiation or virion assembly but is necessary for vector transmission and virus circulation in plants [
23,
24].
In general, the
Polerovirus genus is poorly understood in terms of variation and conservation. A number of proteins within the genus have been identified as multifunctional proteins that are typically highly disordered and hypervariable [
25]. It has been demonstrated that P0 and the RTD of P3–P5 appear to be hypervariable regions and are likely viral species determinants, whereas P2 and P3 are the most genetically stable cistrons in the
Polerovirus genome.
The error-prone nature of RNA-dependent RNA polymerase (RdRp) creates a significant potential for genetic variation in RNA plant viruses, including TuYV [
11,
26]. Mutation and recombination are the two most common types of errors that are thought to cause genetic variation. Evolutionary factors such as genetic drift and selection, including selection pressures associated with virus-vector selection, host plant selection, and maintaining functional structures, could all influence genetic variation [
27]. Several molecular studies have demonstrated significant levels of genetic diversity within TuYV isolates infecting different crops, including oilseed rape, pulses, beetroot etc. [
3,
14,
28,
29,
30]. Furthermore, P0 and P3 play a key role in TuYV virulence [
17,
31]. Therefore, the mode of evolution of these cistrons can reveal information about the epidemiological dynamics of TuYV.
TuYV has a wide host range that includes numerous crops and weed species from different plant families like
Brassicaceae,
Fabaceae,
Chenopodiaceae, and
Compositae [
15,
32,
33,
34,
35,
36]. The wide host range increases the potential reservoirs of TuYV inoculum and acts as a “green bridge” for both virus and its aphid vectors [
37]. The diverse range of hosts and vectors are the main factors aiding the worldwide distribution of TuYV. Transmission of TuYV occurs by aphids in a persistent, circulative manner, with varying degrees of efficiency [
38,
39].
Myzus persicae (green peach aphid) is often considered the most important vector due to its abundance, wide host range and transmission efficiency [
39].
The objective of this study was to determine the prevalence, incidence, and genetic diversity of polerovirus species associated with pea crops and surrounding putative weed or pasture virus reservoir hosts in Tasmania. The outcome of this study will provide Tasmanian pea growers with a clearer understanding of what viruses may pose a threat to their crops, their putative inoculum sources and whether those potential pathogens may require management.
4. Discussion
In this study, we undertook a comprehensive survey of Tasmanian pea crops and neighbouring weed populations that may be potential virus reservoirs using RT-PCR and NGS approaches. Our findings will facilitate a broader understanding of the distribution, diversity and genomic variability of viral populations associated with pea cropping in Tasmania.
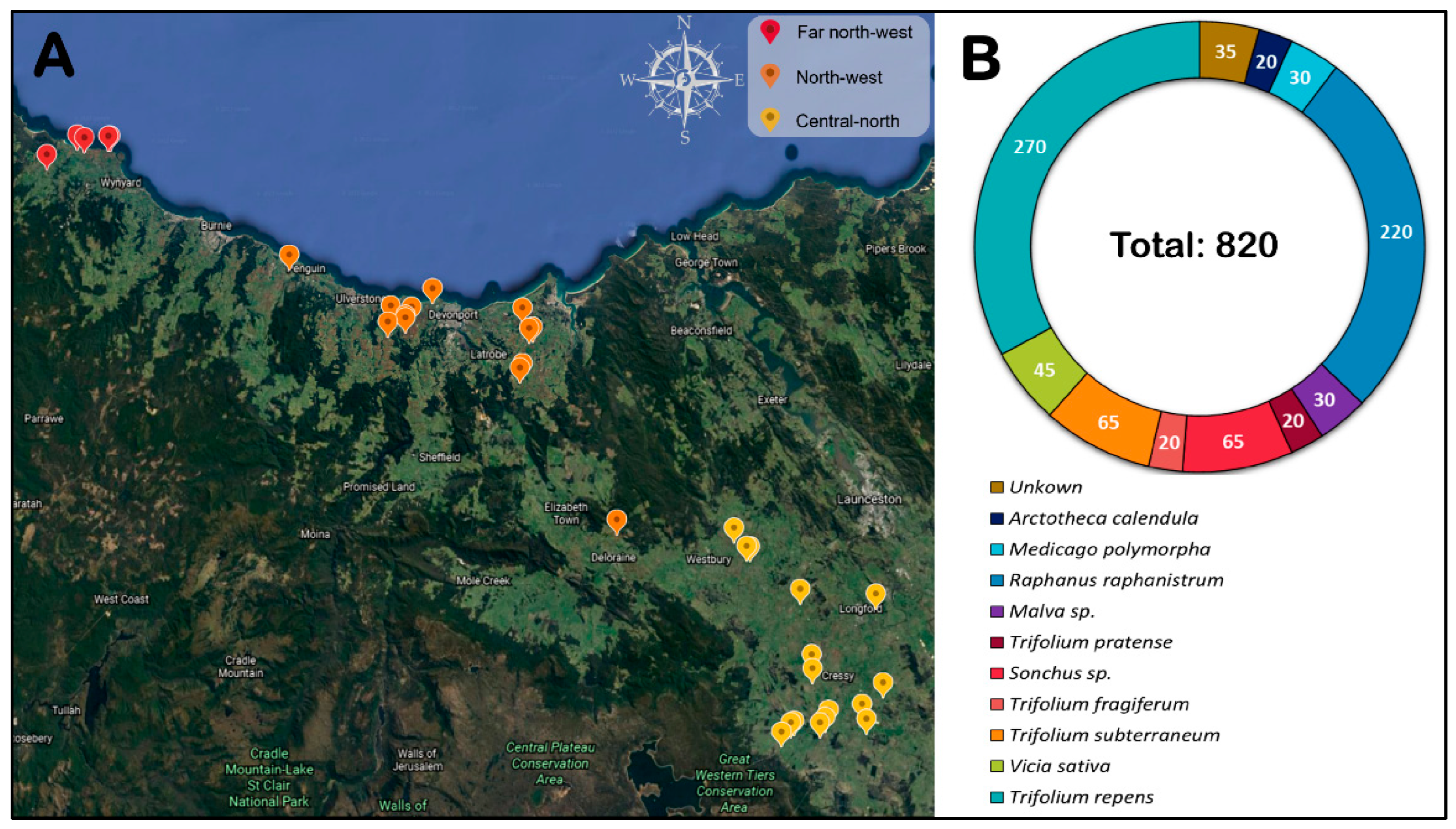
We showed that TuYV is the most prevalent and widely distributed Polerovirus in all surveyed regions, with virus infection levels in crops ranging between 0 and 27.5% of tested plants. The virus was found in 18 of the 22 pea fields located in all three cropping regions. This finding corroborates an earlier report of TuYV incidence in Tasmanian pea crops [
3], where polerovirus (predominantly TuYV) infections were detected in pea crops across three seasons. However, in contrast to the data presented in this manuscript, Polerovirus prevalence did not exceed 60% of surveyed crops, and incidence did not exceed 6.7% in any field in the prior survey. Within the weed hosts neighbouring pea crops, we found that the TuYV infections were greater in
R. raphanistrum and
A. calendula, followed by
T. repens,
Vicia sativa,
Trifolium fragiferum, and
Sonchus sp. TuYV is known to have a wide host range that includes weeds, legume pasture species and other plants from the families
Brassicaceae,
Fabaceae,
Amaranthaceae and
Asteraceae [
14,
33]. It has also been reported across Australia to infect pulse crops (including peas), canola and various other weed species on a regular basis [
14,
36]. A further distinction from the previous Tasmanian survey was a failure to confirm the presence of the newly described PBMYV [
63] in Tasmanian pea crops, which was previously found at low infection levels corresponding to 9 of 28 polerovirus infections detected [
3].
The rate of TuYV infection varied according to the type of host plant and the location of the sampling sites. However, sampling bias must be considered when discussing the distribution of TuYV and the variation in its infection rate across plant species. We hypothesise that several other factors might also be involved in the variable incidence of the virus, such as stage of infection (early or late), types of aphid vectors and their population dynamics, crop management practices, climatic conditions, etc. However, additional studies are imperative to better understand the specific impact of these factors on TuYV incidence.
In addition, our NGS data confirmed several other viral reads, including an almost full genome of viruses representing five different genera and one unassigned virus. The genus Polerovirus and Potyvirus were each represented by two species members, followed by one member from each of
Luteovirus,
Potexvirus, and
Carlavirus. The unclassified virus was from the family
Partitiviridae (
Table 5). However, except for BrYV, RT-PCR was not performed to further confirm the presence of the viruses detected by NGS [
62]. Notably, three of these viruses (BrYV, RCVMV and RsCV) represent previously undescribed viruses in Tasmania [
4].
BrYV belongs to the genus
Polerovirus and is closely related to but appears to be distinctive from TuYV in terms of the P0 and P5 cistrons sequences [
64,
65,
66]. It has been reported in Asia [
64,
65,
66], and a recent report from Australia described the concatenated ORF-based phylogenetic analysis of collected isolates and demonstrated the paraphyletic relation between TuYV and BrYV [
14]. The host range of BrYV is similar to that of TuYV [
67]. However, BrYV isolates have also been subdivided into three genotypes (BrYV-A, -B, and -C) based on sequence similarity and phylogeny, with greater divergence seen in P0, P1 and P2 than in P3, P4 and P5 [
65,
66].
RCVMV is aphid-transmissible in a non-persistent manner; it can also be transmitted mechanically and by seed [
68,
69,
70,
71,
72]. It was first reported in the United States (USA) [
70] and then more widely [
73,
74,
75,
76], most recently in New Zealand [
77]. Depending on the host species and virus isolate, RCVMV can cause vein mosaic, vein chlorosis, and plant stunting, as well as latent infections [
73,
76,
78,
79]. Though RCVMV has been shown to cause leaf chlorosis and yield loss in susceptible varieties of
Trifolium pratense [
80], assessment of the potential impact of RCVMV on clover requires further studies. That is because other viruses (e.g., AMV,
Bean yellow mosaic virus (BYMV), ClYVV, or WClMV, all present in Tasmania) also naturally infect clover and are often found in mixed infections [
77,
81]. Although the presence of RCVMV in Tasmania has been confirmed (unpublished data), future surveys of legume and pasture crops should be conducted to determine its prevalence.
RsCV is an unclassified member of the family
Partitiviridae, and members of this family are known to infect their hosts asymptomatically, including fungi, plants, some protozoa, and possibly some higher animals [
82,
83,
84]. Members have two essential, double-stranded RNA genome segments ranging from 1.4 to 3.0 kb with RNA1 encoding RdRp, while RNA2 encodes CP [
82]. Among those, plant-infecting partitiviruses are commonly referred to as cryptoviruses. Their host range includes radish, alfalfa, beet, broad bean, carrot,
Brassica spp., white clover, red clover, rose, carnation, hop trefoil, Italian ryegrass, meadow fescue, spinach pear, and pine [
84]. They are seed-transmitted with high efficiency, but there is no systemic infection or cell-to-cell moment due to the lack of a movement protein, so they move vertically through their host’s cell division processes instead [
85].
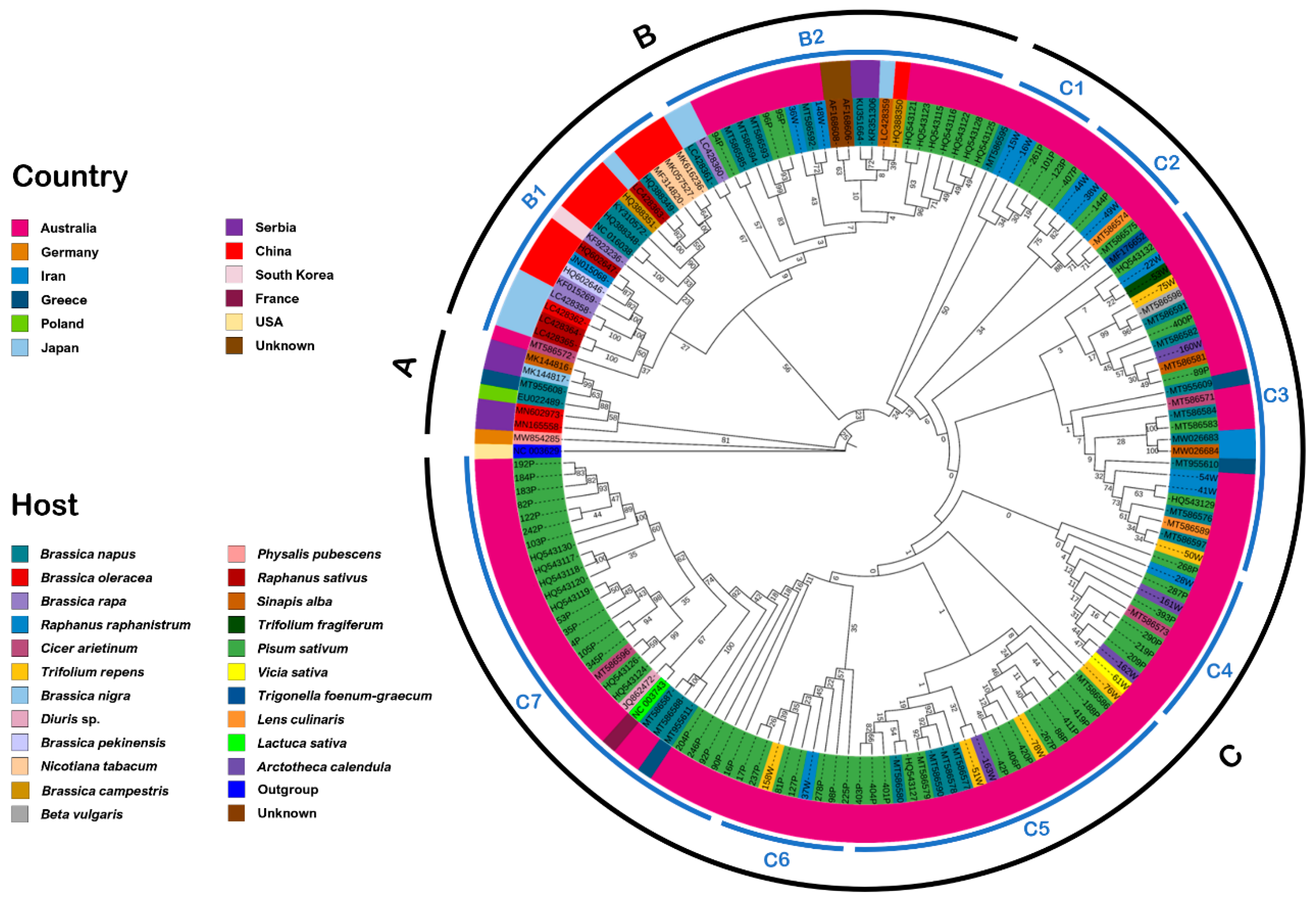
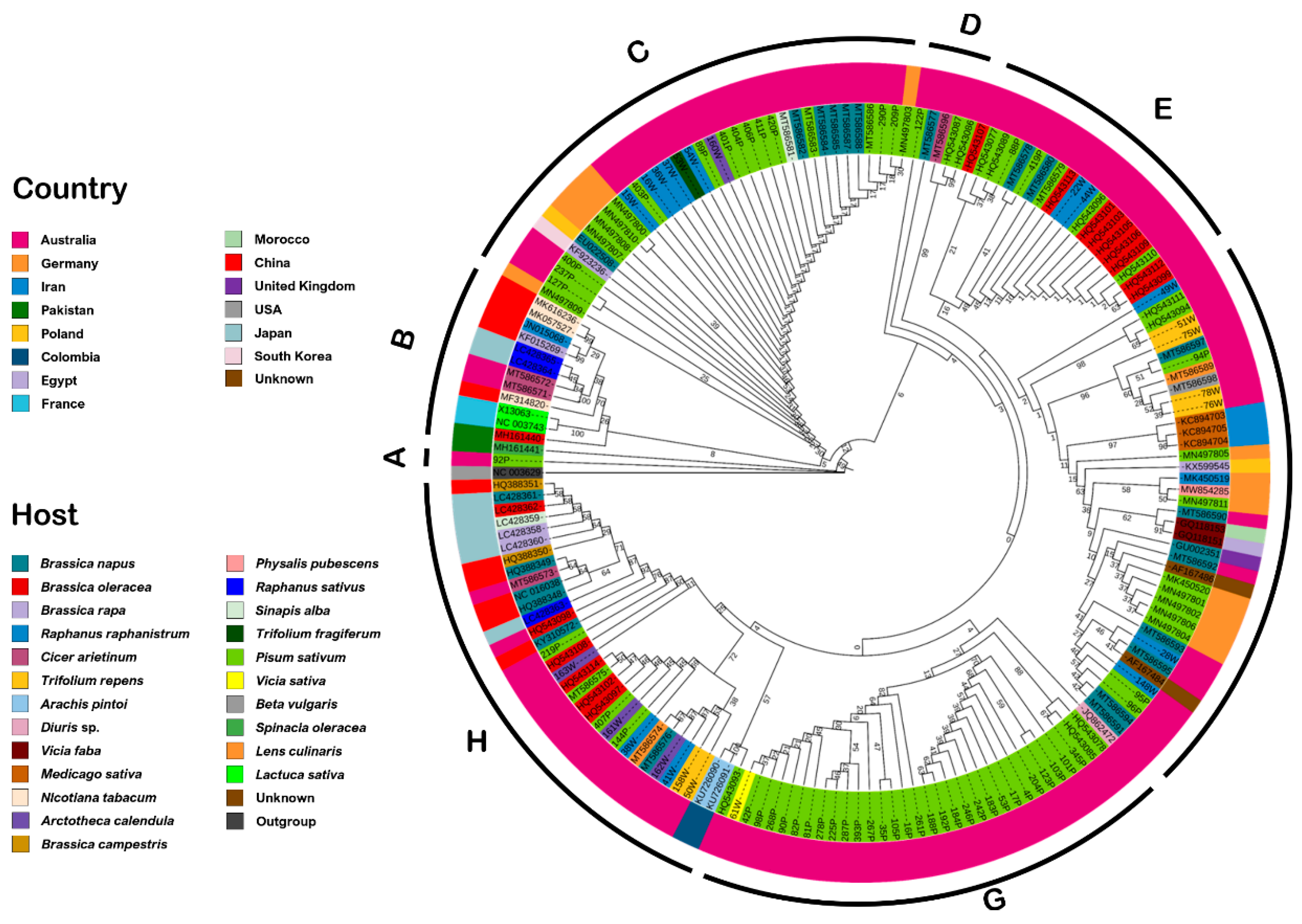
In order to determine the genetic variability of Tasmanian isolates identified by TuYV species-specific PCR, two distinct segments in the genome were sequenced: ORF0, which codes for a protein (P0) involved in post-transcriptional gene silencing, symptom expression, and host range specificity [
86,
87], and ORF3, which codes for the coat protein (P3) [
88,
89]. Seventy-five Tasmanian isolates shared nucleotide identities ranging from 83.2–100% for P0 and from 91.2–100% for P3. Furthermore, the P0 and P3 sequences of the Tasmanian isolates shared 84.6–100% and 89.3–100% nucleotide identities with already published TuYV isolates from GenBank. These results are consistent with previous studies of genetic variation in the
P0 and
P3 genes from the UK [
26,
87]. One of the findings showed that the isolates shared 91.7–100% nucleotide identity for P0 and 94–100% for the P3 gene. The range of nucleotide identities was 86.9–98.8% and 93.5–99.8% for the
P0 and
P3 genes, respectively, when compared with the published isolates from GenBank [
26]. In another study, the European TuYV isolates shared 81.1–100% of the
P0 gene and 90.6–100% nucleotide identities [
87]. The genetic variation of these ORFs indicated that the genomes of polerovirus within these regions are more diverse than previously assumed. When isolates identified as BrYV were included in the phylogenies, we found evidence for discrimination of TuYV and BrYV isolates within the
P0 gene sequences, but no such clear differentiation was found with P3 sequences. Of the new Tasmanian isolates tested, five appeared to cluster with BrYV rather than TuYV in the P0 analysis and may be considered variants of this virus. Notably, subsequent testing of these variant isolates by BrYV species-specific PCR [
90] did not result in positive amplification. Whilst evidence for the separation of BrYV and TuYV as distinct species exists from this and prior studies [
62,
64,
65,
66], suggestions that these viruses may represent a single highly variable species have been made [
14]. Further whole genome sequencing of diverse isolates would be valuable to test these two propositions.
Phylogenetic analysis also highlighted similarities in sequence identity between isolates obtained from weed plants in the vicinity of pea crops and those found within the crops themselves (
Figure 2 and
Figure 3), pointing to the likelihood of virus exchanges between these wild and agricultural plant populations. Additional experimental analyses, including vector and virus monitoring, are required to determine the transmission rate and pattern.
Recombination is more common among RNA viruses than DNA and is a major driving force that greatly contributes to the evolution of viral populations [
91]. Recombination governs genomic diversity and facilitates viral adaptation to varying environments (new host and environmental adaptation), ultimately resulting in the emergence of new/resistant-breaking/virulent variants or strains [
92,
93]. As previously proposed, poleroviruses tend to show higher recombination rates that further contribute to the emergence of new species and their evolution [
94]. The evolution of the genus
Polerovirus is marked by both intraspecific, homologous and interspecific, non-homologous recombination [
14,
95], and generally, in
Luteoviridae, the recombination breakpoints are often at the boundaries of the gene rather than within the gene [
96]. This implies that recombination events in the genome do not occur randomly; rather, these are associated with specific hotspots in the viral genome. Our findings suggested that recombination could be important and may play an important role in the evolution of these viruses. Interestingly, one isolate identified as a BrYV variant based on P0 sequence analysis was subsequently identified as a putative recombinant isolate in recombination analysis. This may suggest genetic exchange between these viruses species may be occurring. We found that recombination breakpoints were detected among 5/75 and 34/75 sequences of
P0 and
P3 genes, respectively (
Table 6), which strongly suggests that the
P0 and
P3 genes have distinct evolutionary histories. Additionally, the considerable phylogenetic incongruence in
P0 and
P3 found in our work supports the concept that recombination might have played a role in the evolution of TuYV. Notably, recombination in genes or mutation in these proteins can affect the biological functions of the viral proteins. For instance, previous studies suggest that host range of different TuYV isolates may be influenced by genetic variation within the
P0 gene [
26,
87]. On the other hand, P3 has a biologically active region that plays a vital role in CP subunit interactions, plant–virus interactions and aphid–virus recognition. Furthermore, the viral particle’s assembly is required for vector transmission and determines which insect vectors can transmit the virus [
97,
98,
99]. Thus, recombination and mutation might affect critical biological functions governed by the
P3 gene. The
P5 readthrough component of the capsid protein has been shown to be highly variable within sequenced TuYV species and across polerovirus species [
87,
100]. The biological significance of variation in the
P5 gene remains unclear at this stage but could be associated with specificity to different vector species. We did not include the analyses of the
P5 gene in this study, which should be considered in future research to acquire a more in-depth understanding of the recombination-driven genome variation.
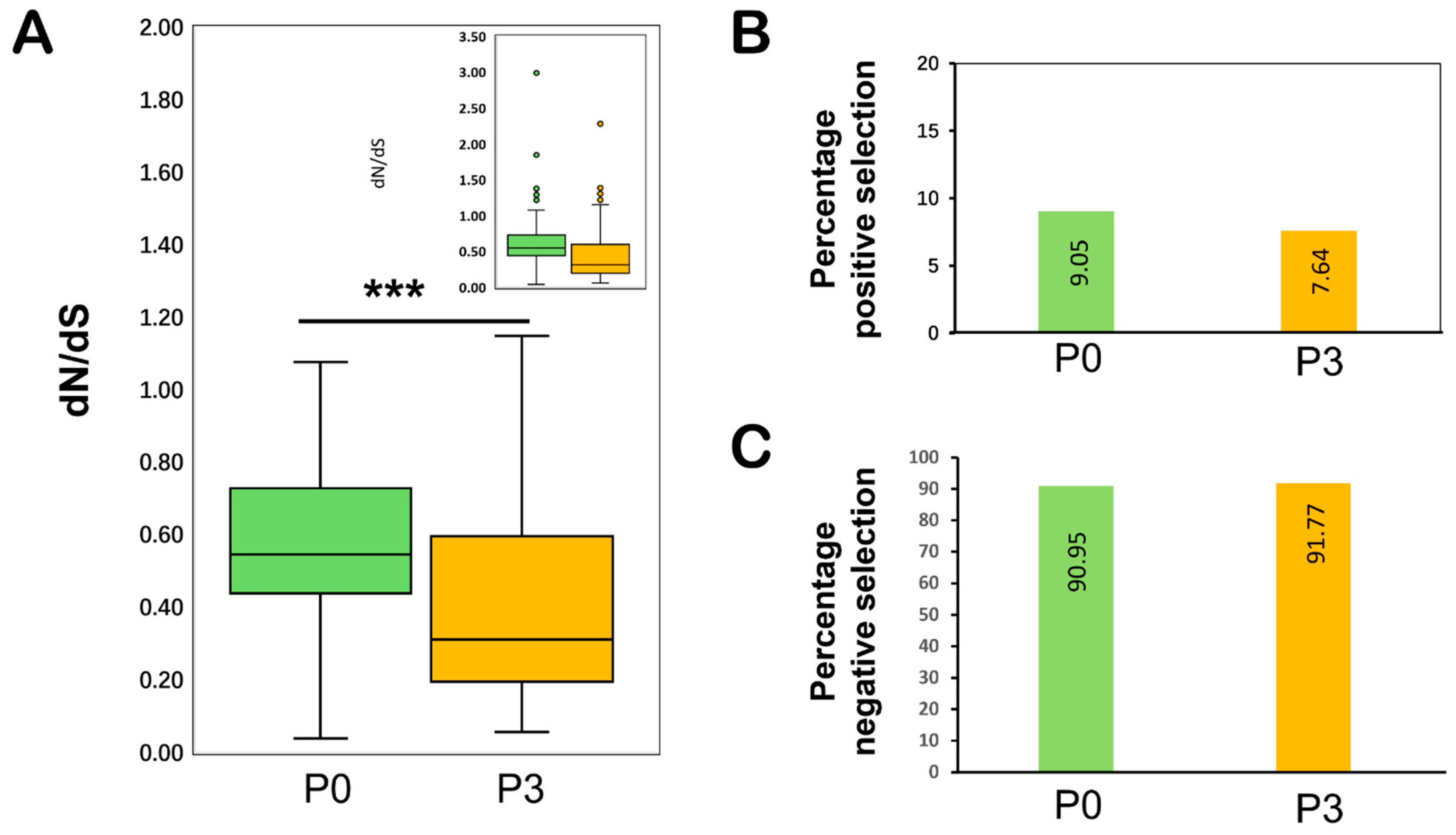
Our results showed that both
P0 and P3 are primarily evolving under purifying selection pressure. Our findings revealed that with an average dN/dS ratio of <1, the majority of the codons remained under negative selection (
Figure 5 and
Table 8), and the overall contribution of negatively selected sites remained >90% (91% for
P0 and 92.4% for
P3). This is consistent with the previous research that concluded that both genes of TuYV (
P0 and
P3) were evolving under strong negative selection pressure [
26]. Negative selection in the TuYV genome, which is required to keep the encoded protein functional (as in P3 and P0 of TuYV), may have helped to eliminate deleterious variants. TuYV’s
P0 gene is involved in RNA-silencing suppression [
17], and mutations in this gene are expected to pose a significant impact on virus fitness, limiting genetic diversity and thus influencing dN/dS ratio estimates [
101,
102].
Taken together, this study advances our current understanding of the genetically diversified and evolving populations of TuYV infecting Tasmanian pea crops. Our findings also provide a practical framework necessary for understanding the current diversity and distribution of poleroviruses in three selected regions of Tasmania. It also provides important information on epidemiological aspects and management of viral diseases of pea crops. Future studies based on the full genome-based analyses of the genetic variations will expand our understanding of the evolutionary patterns existing among TuYV populations in Tasmania.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}