
Enteroviral 2B Interacts with VDAC3 to Regulate Reactive Oxygen Species Generation That Is Essential to Viral Replication

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. Molecular Biology Techniques
2.3. Virological Techniques
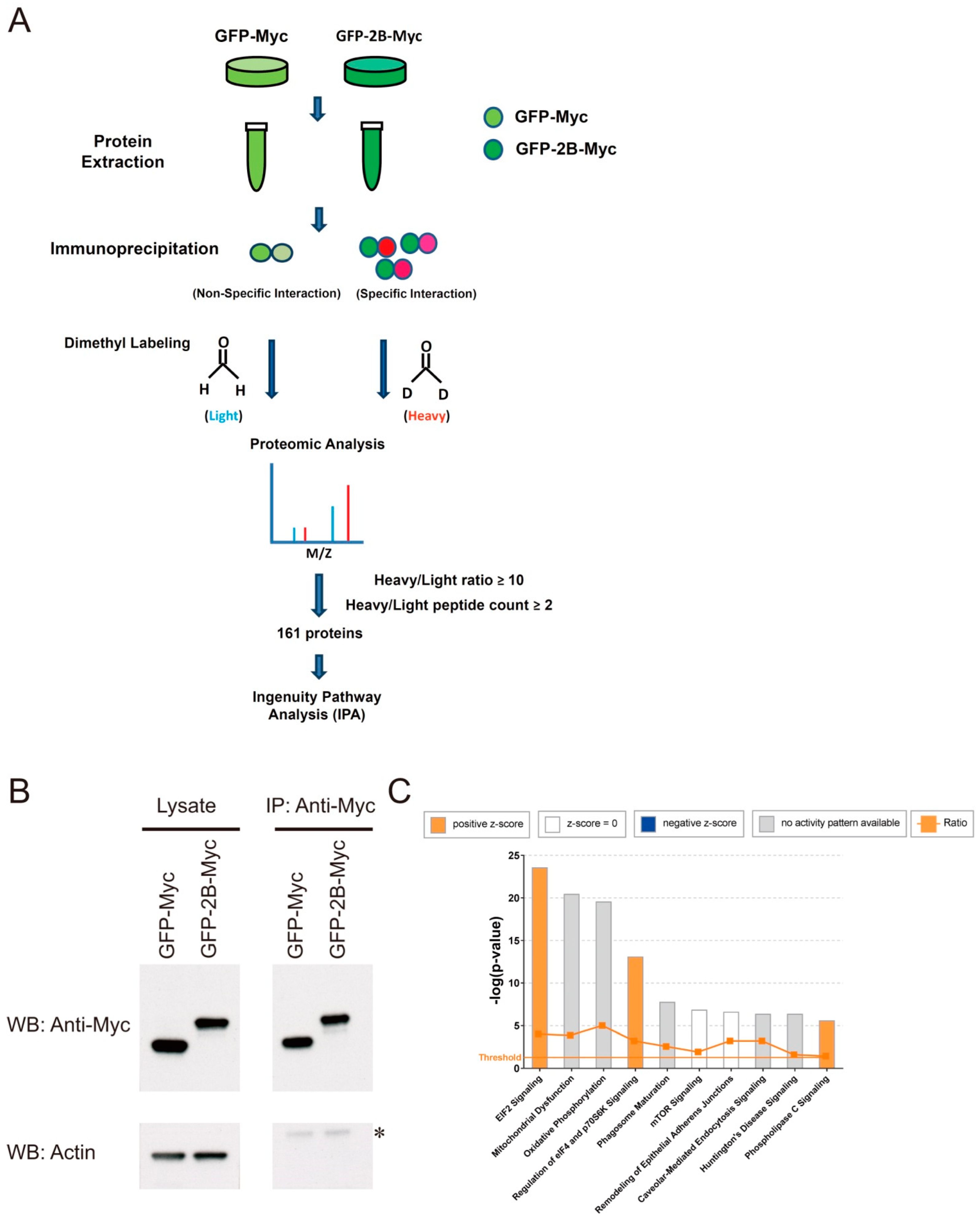
2.4. Immunoprecipitation and Proteomic Analysis
2.5. Immunological Techniques
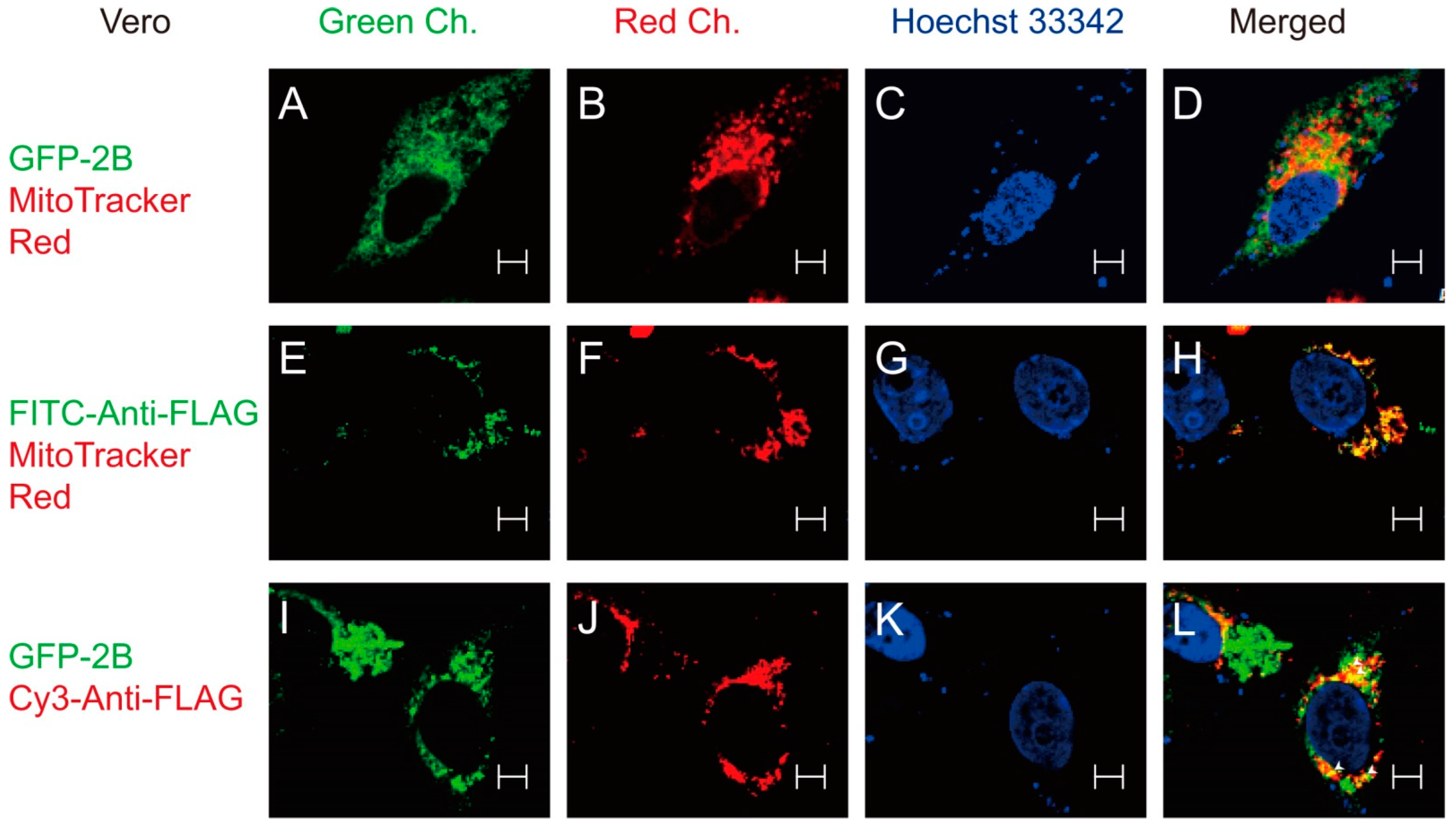
2.6. Confocal Microscopy
2.7. Cytometric Analysis
2.8. Metabolite Analysis
2.9. Statistical Analysis
3. Results
3.1. Reduction in EV71-Induced ROS Generation Is Inhibitory to Viral Replication
3.2. Identification of Protein-Protein Interaction That Is Important in Mitochondrial ROS Generation
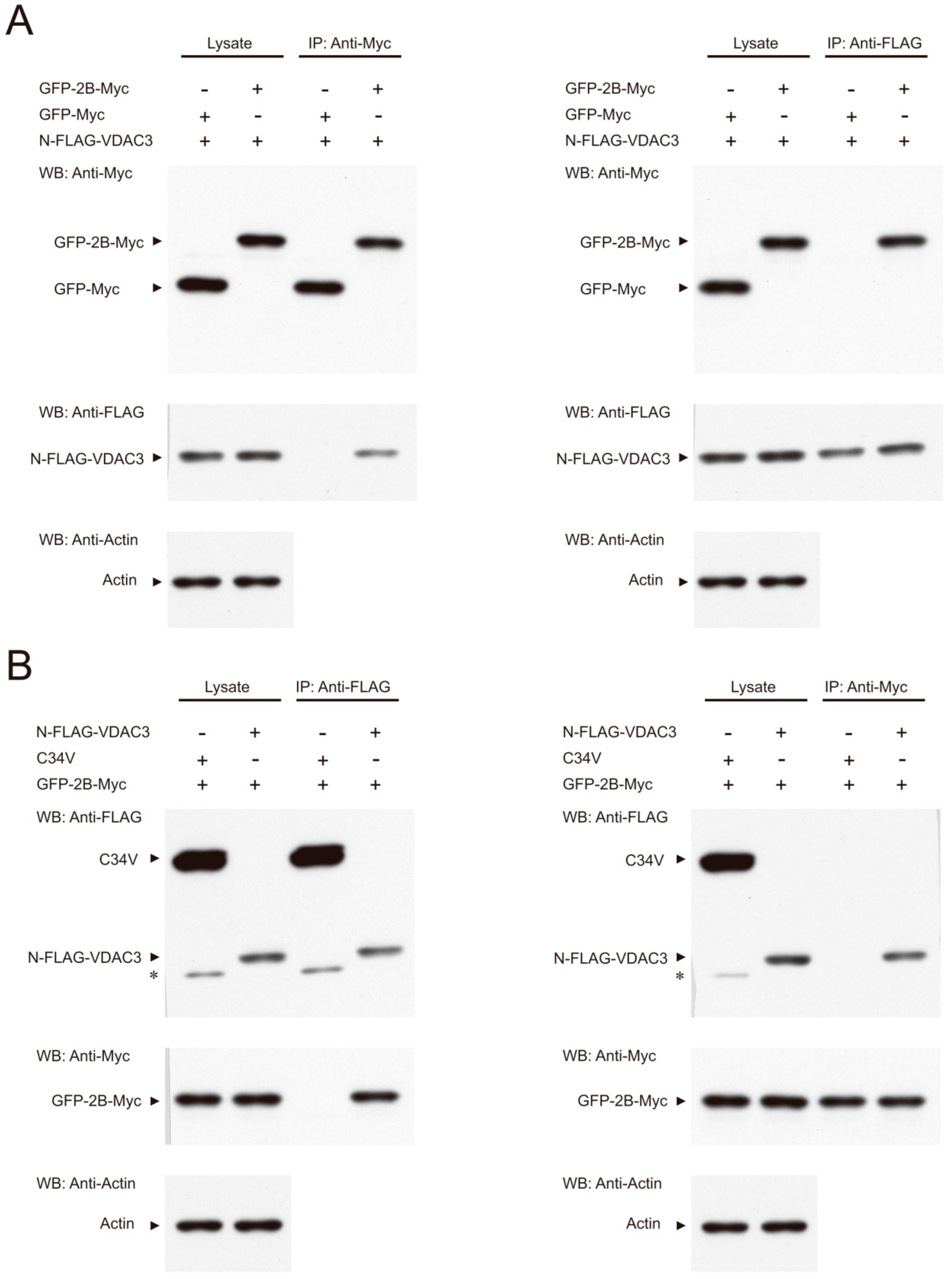
3.3. Interaction between 2B and VDAC3
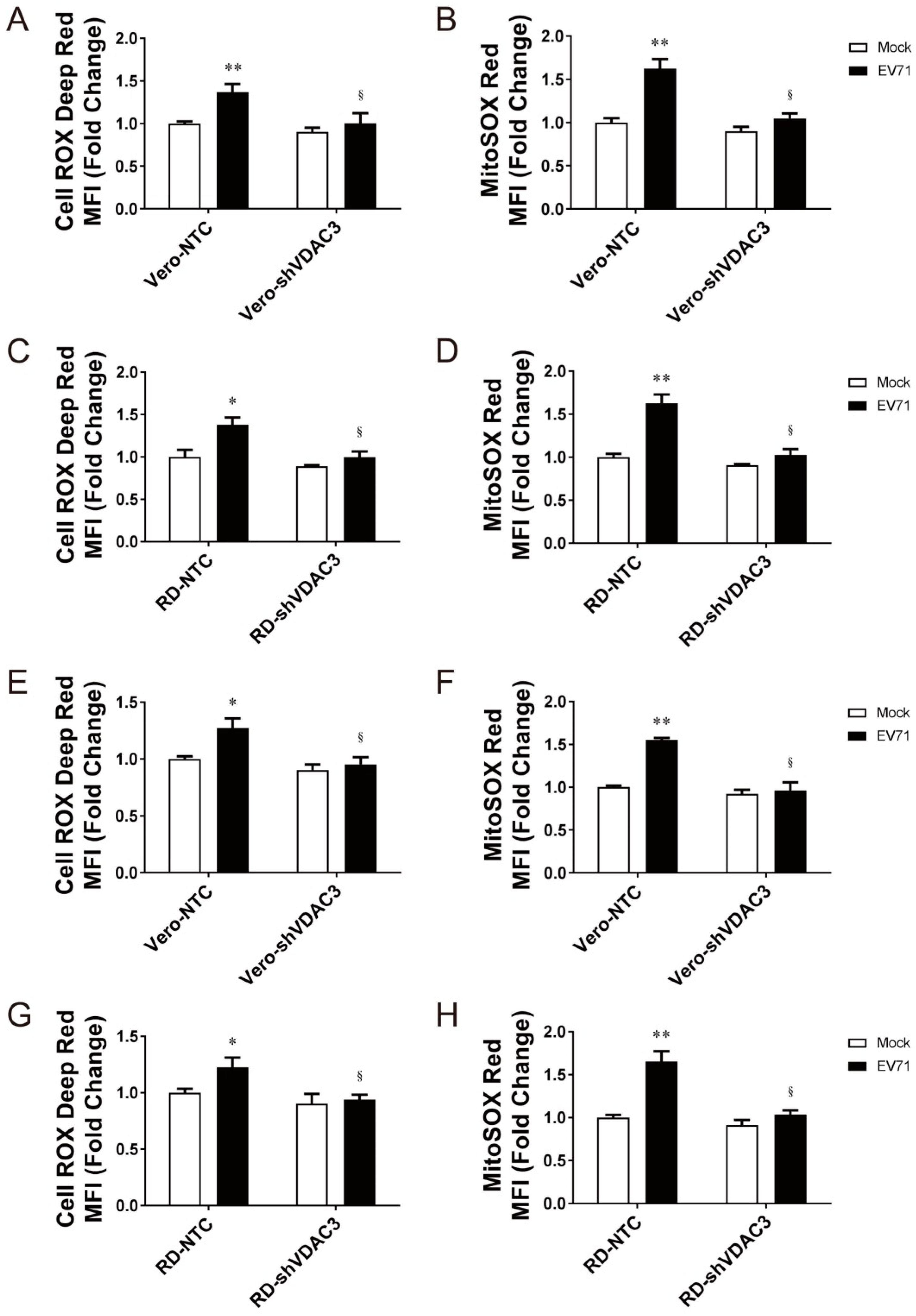
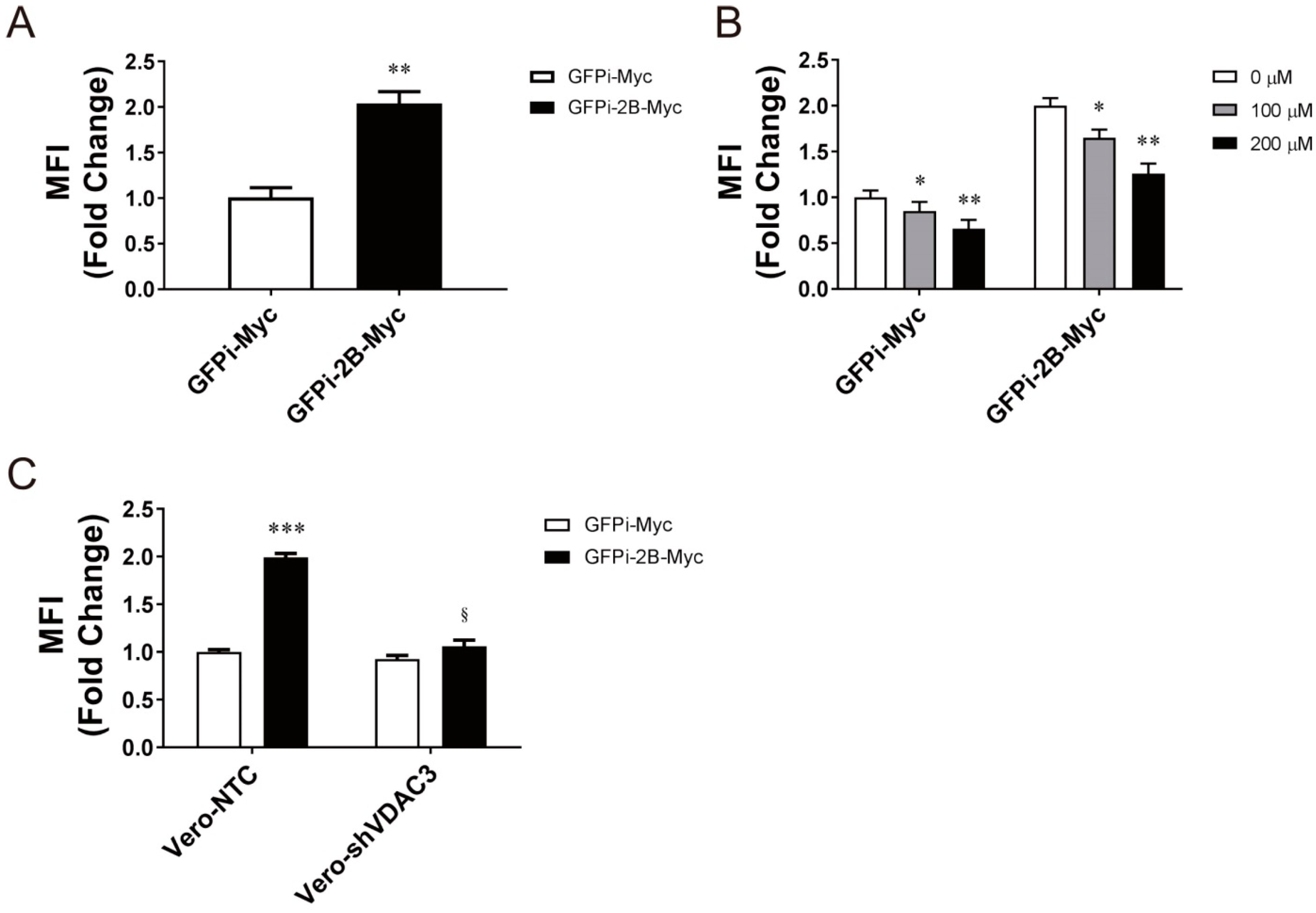
3.4. Specific Role of VDAC3 in EV71 Replication
3.5. Interaction between 2B and VDAC3 in EV71-Induced Mitochondrial ROS Generation
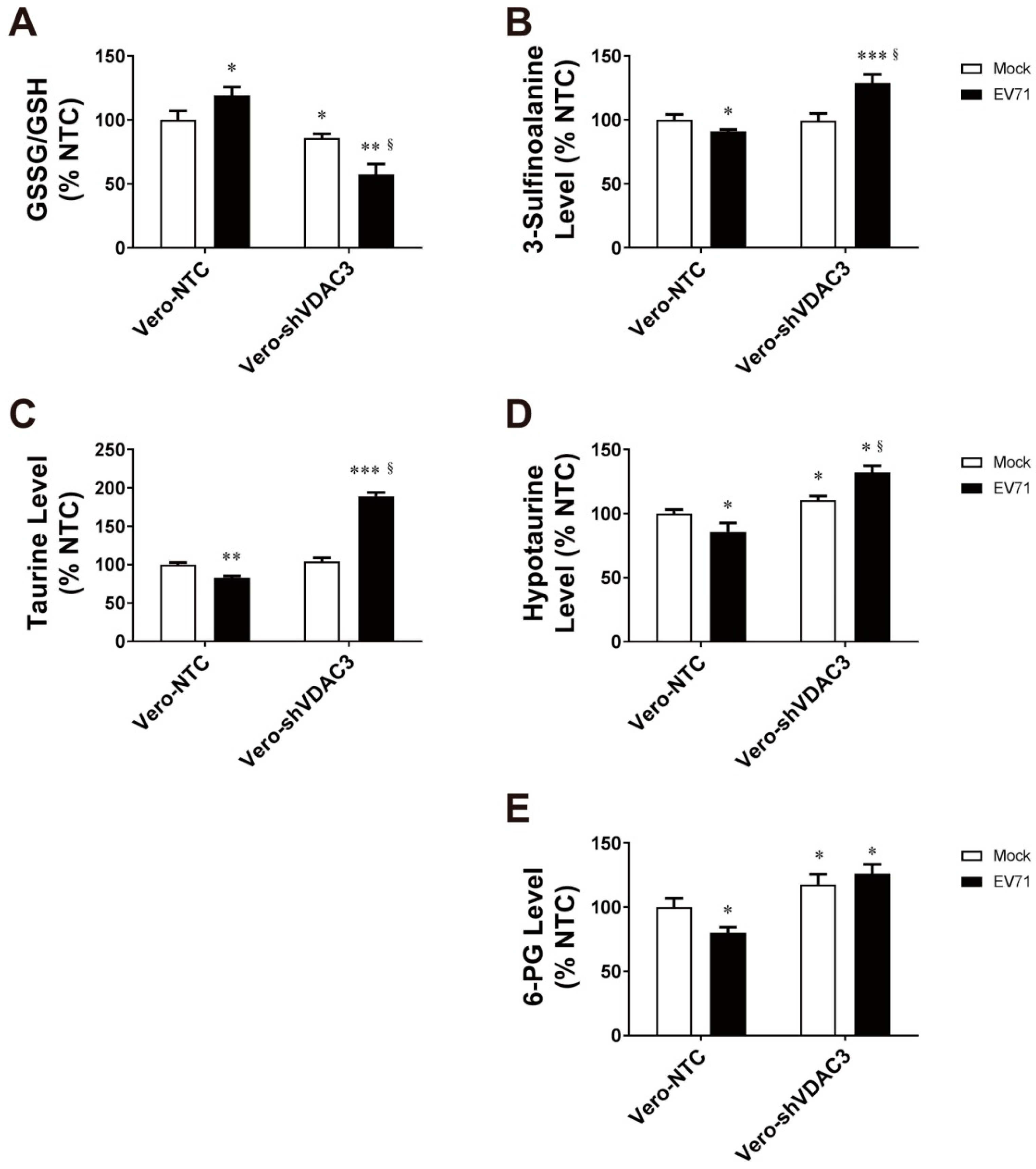
3.6. EV71 Infection Is Associated with Changes in Antioxidant Metabolism
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulou, Z.; Pliaka, V.; Amoutzias, G.; Markoulatos, P. Recombination among human non-polio enteroviruses: Implications for epidemiology and evolution. Virus Genes 2015, 50, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, M.H.; Wong, S.C.; Lewthwaite, P.; Cardosa, M.J.; Solomon, T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010, 9, 1097–1105. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, T.; Hu, Y.; Wang, X.; Du, J.; Li, Y.; Sun, S.; Sun, X.; Li, Z.; Jin, Q. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol. J. 2011, 8, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.-X.; Wu, B.; Bao, W.-X.; Han, F.-A.; Xu, L.; Ge, Q.-J.; Yang, J.; Yuan, Z.-H.; Miao, C.-H.; Huang, X.-X.; et al. Epidemiology of hand, foot, and mouth disease and genotype characterization of Enterovirus 71 in Jiangsu, China. J. Clin. Virol. 2010, 49, 100–104. [Google Scholar] [CrossRef]
- Cheng, M.-L.; Chien, K.-Y.; Lai, C.-H.; Li, G.-J.; Lin, J.-F.; Ho, H.-Y. Metabolic Reprogramming of Host Cells in Response to Enteroviral Infection. Cells 2020, 9, 473. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; LaCount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Ho, H.-Y.; Cheng, M.-L.; Weng, S.-F.; Chang, L.; Yeh, T.-T.; Shih, S.-R.; Chiu, D.T.-Y. Glucose-6-phosphate dehydrogenase deficiency enhances enterovirus 71 infection. J. Gen. Virol. 2008, 89, 2080–2089. [Google Scholar] [CrossRef]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2- generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, F.J.; Roederer, M.; Herzenberg, L.A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1990, 87, 9943–9947. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E. Sendai virus stimulates chemiluminescence in mouse spleen cells. Biochem. Biophys. Res. Commun. 1979, 91, 383–392. [Google Scholar] [CrossRef]
- Martinez, I.; García-Carpizo, V.; Guijarro, T.; García-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 7, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Biagioli, M.; Kaul, P.; Singh, I.; Turner, R.B. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic. Biol. Med. 1999, 26, 454–462. [Google Scholar] [CrossRef]
- Lim, J.-Y.; Oh, E.; Kim, Y.; Jung, W.-W.; Kim, H.-S.; Lee, J.; Sul, D. Enhanced oxidative damage to DNA, lipids, and proteins and levels of some antioxidant enzymes, cytokines, and heat shock proteins in patients infected with influenza H1N1 virus. Acta Virol. 2014, 58, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.P.; Lee, J.C.-Y.; Loke, W.M.; Yeo, L.; Quek, A.M.; Lim, E.C.; Halliwell, B.; Seet, R.C.-S. Does Influenza A Infection Increase Oxidative Damage? Antioxid. Redox Signal. 2014, 21, 1025–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, A.B.; Muthuraman, K.R.; Mariappan, V.; Belur, S.S.; Lokesh, S.; Rajendiran, S. Oxidative stress response in the pathogenesis of dengue virus virulence, disease prognosis and therapeutics: An update. Arch. Virol. 2019, 164, 2895–2908. [Google Scholar] [CrossRef]
- Cheng, M.-L.; Weng, S.-F.; Kuo, C.-H.; Ho, H.-Y. Enterovirus 71 Induces Mitochondrial Reactive Oxygen Species Generation That is Required for Efficient Replication. PLoS ONE 2014, 9, e113234. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Osahon, O.; Vides, D.B.; Hanania, N.; Minard, C.G.; Sekhar, R.V. Severe Glutathione Deficiency, Oxidative Stress and Oxidant Damage in Adults Hospitalized with COVID-19: Implications for GlyNAC (Glycine and N-Acetylcysteine) Supplementation. Antioxidants 2021, 11, 50. [Google Scholar] [CrossRef]
- Piccoli, C.; Scrima, R.; Quarato, G.; D’Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef]
- Macho, A.; Calzado, M.A.; Jimenez-Reina, L.; Ceballos, E.; Leon, J.; Munoz, E. Susceptibility of HIV-1-TAT transfected cells to undergo apoptosis. Biochemical mechanisms. Oncogene 1999, 18, 7543–7551. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Reina, S.; Checchetto, V. Voltage-Dependent Anion Selective Channel 3: Unraveling Structural and Functional Features of the Least Known Porin Isoform. Front. Physiol. 2021, 12, 784867. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, A.; Sheiko, T.; Graham, B.H.; Craigen, W.J. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. Biochim. Biophys. Acta 2012, 1818, 1477–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najbauer, E.E.; Becker, S.; Giller, K.; Zweckstetter, M.; Lange, A.; Steinem, C.; de Groot, B.L.; Griesinger, C.; Andreas, L.B. Structure, gating and interactions of the voltage-dependent anion channel. Eur. Biophys. J. 2021, 50, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Ao, D.; Sun, S.-Q.; Guo, H.-C. Topology and biological function of enterovirus non-structural protein 2B as a member of the viroporin family. Vet. Res. 2014, 45, 87. [Google Scholar] [CrossRef] [PubMed]
- Agirre, A.; Lorizate, M.; Nir, S.; Nieva, J.L. Poliovirus 2b insertion into lipid monolayers and pore formation in vesicles modulated by anionic phospholipids. Biochim. Biophys. Acta 2008, 1778, 2621–2626. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Wang, K.; Yu, W.; Lu, W.; Xu, K.; Wang, J.; Ye, B.; Schwarz, W.; Jin, Q.; Sun, B. DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res. 2011, 21, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.; de Jong, A.S.; Lanke, K.W.H.; Melchers, W.; Willems, P.H.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J.M. The Coxsackievirus 2B Protein Suppresses Apoptotic Host Cell Responses by Manipulating Intracellular Ca2+ Homeostasis. J. Biol. Chem. 2004, 279, 18440–18450. [Google Scholar] [CrossRef] [Green Version]
- Supasorn, O.; Tongtawe, P.; Srimanote, P.; Rattanakomol, P.; Thanongsaksrikul, J. A nonstructural 2B protein of enterovirus A71 increases cytosolic Ca2+ and induces apoptosis in human neuroblastoma SH-SY5Y cells. J. Neurovirol. 2020, 26, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Thyrsted, J.; Holm, C.K. Virus-induced metabolic reprogramming and innate sensing hereof by the infected host. Curr. Opin. Biotechnol. 2021, 68, 44–50. [Google Scholar] [CrossRef]
- Boya, P.; de la Peña, A.; Beloqui, O.; Larrea, E.; Conchillo, M.; Castelruiz, Y.; Civeira, M.-P.; Prieto, J. Antioxidant status and glutathione metabolism in peripheral blood mononuclear cells from patients with chronic hepatitis C. J. Hepatol. 1999, 31, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-G.; Leu, Y.-L.; Cheng, M.-L.; Ting, S.C.; Liu, C.-C.; Wang, S.-D.; Yang, C.-H.; Hung, C.-Y.; Sakurai, H.; Chen, K.-H.; et al. Anti-enterovirus 71 activities of Melissa officinalis extract and its biologically active constituent rosmarinic acid. Sci. Rep. 2017, 7, 12264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, P.X.; Panda, D.; Das, P.B.; Das, S.C.; Das, A.; Pattnaik, A.K. A single amino acid change resulting in loss of fluorescence of eGFP in a viral fusion protein confers fitness and growth advantage to the recombinant vesicular stomatitis virus. Virology 2012, 432, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.; Puhl, H.L., 3rd; Ikeda, S.R. Rapid Modification of Proteins Using a Rapamycin-Inducible Tobacco Etch Virus Protease System. PLoS ONE 2009, 4, e7474. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-H.; Liang, C.-T.; Jiang, S.-T.; Chen, K.-H.; Yang, C.-C.; Cheng, M.-L.; Ho, H.-Y. A Novel Murine Model Expressing a Chimeric mSCARB2/hSCARB2 Receptor Is Highly Susceptible to Oral Infection with Clinical Isolates of Enterovirus 71. J. Virol. 2019, 93, e00183-19. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-G.; Cheng, M.-L.; Chen, K.-H.; Horng, J.-T.; Liu, C.-C.; Wang, S.-M.; Sakurai, H.; Leu, Y.-L.; Wang, S.-D.; Ho, H.-Y. Antiviral activities of Schizonepeta tenuifolia Briq. against enterovirus 71 in vitro and in vivo. Sci. Rep. 2017, 7, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cerqueira Cesar, M.; Wilson, J.E. All three isoforms of the voltage-dependent anion channel (VDAC1, VDAC2, and VDAC3) are present in mitochondria from bovine, rabbit, and rat brain. Arch. Biochem. Biophys. 2004, 422, 191–196. [Google Scholar] [CrossRef]
- Kim, B.-H.; Oh, J.-M.; Yun, K.-U.; Kim, C.-H.; Kim, S.-K. Methods for Evaluation of Antioxidant Activity: Application to Taurine. Toxicol. Res. 2007, 23, 263–269. [Google Scholar] [CrossRef]
- Cymerys, J.; Chodkowski, M.; Słońska, A.; Krzyżowska, M.; Bańbura, M.W. Disturbances of mitochondrial dynamics in cultured neurons infected with human herpesvirus type 1 and type 2. J. Neurovirol. 2019, 25, 765–782. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Xiang, H.; Bai, X.; Fei, N.; Huang, Y.; Song, X.; Zhang, H.; Zhang, L.; Tong, D. Porcine parvovirus infection activates mitochondria-mediated apoptotic signaling pathway by inducing ROS accumulation. Virol. J. 2016, 13, 26. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Cheng, Y.; Song, S.; Qiu, J.; Yi, L.; Cao, Z.; Li, J.; Cheng, S.; Wang, J. Viral Nonstructural Protein 1 Induces Mitochondrion-Mediated Apoptosis in Mink Enteritis Virus Infection. J. Virol. 2019, 93, e01249-19. [Google Scholar] [CrossRef]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human Hepatitis B Virus-X Protein Alters Mitochondrial Function and Physiology in Human Liver Cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron Transport and Increases Reactive Oxygen Species (ROS) Production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [Green Version]
- Jassey, A.; Liu, C.-H.; Changou, C.; Richardson, C.; Hsu, H.-Y.; Lin, L.-T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Kammouni, W.; Wood, H.; Saleh, A.; Appolinario, C.M.; Fernyhough, P.; Jackson, A.C. Rabies virus phosphoprotein interacts with mitochondrial Complex I and induces mitochondrial dysfunction and oxidative stress. J. Neurovirol. 2015, 21, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of Host Cell Mitochondria by RSV to Favor Virus Production is Dependent on Inhibition of Mitochondrial Complex I and ROS Generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef] [Green Version]
- Claus, C.; Schönefeld, K.; Hübner, D.; Chey, S.; Reibetanz, U.; Liebert, U.G. Activity Increase in Respiratory Chain Complexes by Rubella Virus with Marginal Induction of Oxidative Stress. J. Virol. 2013, 87, 8481–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zobel, S.; Lorenz, M.; Frascaroli, G.; Böhnke, J.; Bilz, N.C.; Stanifer, M.L.; Boulant, S.; Bergs, S.; Liebert, U.G.; Claus, C. Rubella Virus Strain-Associated Differences in the Induction of Oxidative Stress Are Independent of Their Interferon Activation. Viruses 2018, 10, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuren, L.M.; Prestes, E.B.; Papa, M.P.; de Carvalho, L.R.P.; Mustafá, Y.M.; da Costa, L.S.; Da Poian, A.T.; Bozza, M.T.; Arruda, L.B. Infection of Endothelial Cells by Dengue Virus Induces ROS Production by Different Sources Affecting Virus Replication, Cellular Activation, Death and Vascular Permeability. Front. Immunol. 2022, 13, 810376. [Google Scholar] [CrossRef] [PubMed]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-Like Receptor X-1 Is Required for Rhinovirus-Induced Barrier Dysfunction in Airway Epithelial Cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Tan, C.L.; Gunaratne, J.; Quek, L.S.; Nei, W.L.; Thierry, F.; Bellanger, S. Localization of HPV-18 E2 at Mitochondrial Membranes Induces ROS Release and Modulates Host Cell Metabolism. PLoS ONE 2013, 8, e75625. [Google Scholar] [CrossRef] [Green Version]
- Soucy-Faulkner, A.; Mukawera, E.; Fink, K.; Martel, A.; Jouan, L.; Nzengue, Y.; Lamarre, D.; Vande Velde, C.; Grandvaux, N. Requirement of NOX2 and Reactive Oxygen Species for Efficient RIG-I-Mediated Antiviral Response through Regulation of MAVS Expression. PLOS Pathog. 2010, 6, e1000930. [Google Scholar] [CrossRef] [Green Version]
- Buskiewicz, I.A.; Montgomery, T.; Yasewicz, E.C.; Huber, S.A.; Murphy, M.P.; Hartley, R.C.; Kelly, R.; Crow, M.K.; Perl, A.; Budd, R.C.; et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci. Signal. 2016, 9, ra115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.B.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. eLife 2019, 8, e42448. [Google Scholar] [CrossRef]
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.R.S.; et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69. [Google Scholar] [CrossRef]
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.S.; Drummond, G.; Selemidis, S. Inhibition of Nox2 Oxidase Activity Ameliorates Influenza A Virus-Induced Lung Inflammation. PLOS Pathog. 2011, 7, e1001271. [Google Scholar] [CrossRef] [Green Version]
- To, E.E.; Erlich, J.R.; Liong, F.; Luong, R.; Liong, S.; Esaq, F.; Oseghale, O.; Anthony, D.; McQualter, J.; Bozinovski, S.; et al. Mitochondrial Reactive Oxygen Species Contribute to Pathological Inflammation During Influenza A Virus Infection in Mice. Antioxid. Redox Signal. 2020, 32, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Van Kuppeveld, F.J.; Hoenderop, J.G.; Smeets, R.L.; Willems, P.H.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997, 16, 3519–3532. [Google Scholar] [CrossRef] [PubMed]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, Y.; Castro, I.G.; Schuldiner, M. The Fast and the Furious: Golgi Contact Sites. Contact 2021, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Komarov, A.; Bezrukov, S.M.; Colombini, M. Dynamics of Nucleotides in VDAC Channels: Structure-Specific Noise Generation. Biophys. J. 2002, 82, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Mazure, N.M. VDAC in Cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef]
- Colombini, M. The VDAC channel: Molecular basis for selectivity. Biochim. Biophys. Acta 2016, 1863, 2498–2502. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Maldonado, E.N. VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation. Adv. Cancer Res. 2018, 138, 41–69. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, Z.; Huh, K.-W.; Lasher, R.; Siddiqui, A. Hepatitis B Virus X Protein Colocalizes to Mitochondria with a Human Voltage-Dependent Anion Channel, HVDAC3, and Alters Its Transmembrane Potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heslop, K.A.; Milesi, V.; Maldonado, E.N. VDAC Modulation of Cancer Metabolism: Advances and Therapeutic Challenges. Front. Physiol. 2021, 12, 742839. [Google Scholar] [CrossRef]
- Reina, S.; Nibali, S.C.; Tomasello, M.F.; Magrì, A.; Messina, A.; De Pinto, V. Voltage Dependent Anion Channel 3 (VDAC3) protects mitochondria from oxidative stress. Redox Biol. 2022, 51, 102264. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Reina, S.; Guarino, F.; Magrì, A.; Tomasello, F.; Clark, R.E.; Ramsay, R.R.; De Pinto, V. Live cell interactome of the human voltage dependent anion channel 3 (VDAC3) revealed in HeLa cells by affinity purification tag technique. Mol. BioSyst. 2014, 10, 2134–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.L.; Neumann, C.A. Redoxins as gatekeepers of the transcriptional oxidative stress response. Redox Biol. 2019, 21, 101104. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.L.; Nadler, M.; Skoko, J.J.; Bertomeu, T.; Pelosi, A.; Shafaei, P.M.; Levine, K.; Schempf, A.; Pennarun, B.; Yang, B.; et al. A Peroxidase Peroxiredoxin 1-Specific Redox Regulation of the Novel FOXO3 microRNA Target let-7. Antioxid. Redox Signal. 2018, 28, 62–77. [Google Scholar] [CrossRef]
- Nakai, S.; Oyabu, M.; Hatazawa, Y.; Akashi, S.; Kitamura, T.; Miura, S.; Kamei, Y. FOXO1 suppresses PGC-1β gene expression in skeletal muscles. FEBS Open Bio 2020, 10, 1373–1388. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Pinkaew, D.; Doan, H.Q.; Jacob, R.B.; Verma, S.K.; Friedman, H.; Peterson, A.C.; Kuyumcu-Martinez, M.N.; McDougal, O.M.; Fujise, K. Fortilin potentiates the peroxidase activity of Peroxiredoxin-1 and protects against alcohol-induced liver damage in mice. Sci. Rep. 2016, 6, 18701. [Google Scholar] [CrossRef] [Green Version]
- Stipanuk, M.H. Metabolism of Sulfur-Containing Amino Acids: How the Body Copes with Excess Methionine, Cysteine, and Sulfide. J. Nutr. 2020, 150, 2494S–2505S. [Google Scholar] [CrossRef]
- Jong, C.J.; Sandal, P.; Schaffer, S.W. The Role of Taurine in Mitochondria Health: More than Just an Antioxidant. Molecules 2021, 26, 4913. [Google Scholar] [CrossRef] [PubMed]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of taurine, hypotaurine and their metabolic precursors. Biochem. J. 1988, 256, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Celino, F.T.; Shimizu-Yamaguchi, S.; Miura, C.; Miura, T. Taurine plays an important role in the protection of spermatogonia from oxidative stress. Amino Acids 2012, 43, 2359–2369. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, H.; Rehman, H.; Banerjee, B.D.; Raisuddin, S.; Parvez, S. Attenuation of tamoxifen-induced hepatotoxicity by taurine in mice. Clin. Chim. Acta 2006, 370, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.-Y.; Cheng, M.-L.; Weng, S.-F.; Leu, Y.-L.; Chiu, D.T.-Y. Antiviral Effect of Epigallocatechin Gallate on Enterovirus 71. J. Agric. Food Chem. 2009, 57, 6140–6147. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, R.C.; Steel, J.J.; Moon, S.; Soltani, E.; Geiss, B.J. Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology 2015, 475, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Stankovic-Valentin, N.; Drzewicka, K.; König, C.; Schiebel, E.; Melchior, F. Redox regulation of SUMO enzymes is required for ATM activity and survival in oxidative stress. EMBO J. 2016, 35, 1312–1329. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-C.; Chang, L.-Y.; Wang, Y.-W.; Chen, Y.-C.; Weng, K.-F.; Shih, S.-R.; Shih, H.-M. Sumoylation-promoted Enterovirus 71 3C Degradation Correlates with a Reduction in Viral Replication and Cell Apoptosis. J. Biol. Chem. 2011, 286, 31373–31384. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From Molecular Mechanisms to Health Outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.-L.; Wu, C.-H.; Chien, K.-Y.; Lai, C.-H.; Li, G.-J.; Liu, Y.-Y.; Lin, G.; Ho, H.-Y. Enteroviral 2B Interacts with VDAC3 to Regulate Reactive Oxygen Species Generation That Is Essential to Viral Replication. Viruses 2022, 14, 1717. https://doi.org/10.3390/v14081717
Cheng M-L, Wu C-H, Chien K-Y, Lai C-H, Li G-J, Liu Y-Y, Lin G, Ho H-Y. Enteroviral 2B Interacts with VDAC3 to Regulate Reactive Oxygen Species Generation That Is Essential to Viral Replication. Viruses. 2022; 14(8):1717. https://doi.org/10.3390/v14081717
Chicago/Turabian StyleCheng, Mei-Ling, Chien-Hsiang Wu, Kun-Yi Chien, Chien-Hsueh Lai, Guan-Jie Li, Yuan-Yu Liu, Gigin Lin, and Hung-Yao Ho. 2022. "Enteroviral 2B Interacts with VDAC3 to Regulate Reactive Oxygen Species Generation That Is Essential to Viral Replication" Viruses 14, no. 8: 1717. https://doi.org/10.3390/v14081717