
Spatial Dispersal of Epstein–Barr Virus in South America Reveals an African American Variant in Brazilian Lymphomas
 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. DNA Extraction, Amplification, and Sequencing
2.3. Detection of Polymorphisms, Phylogenetics and Phylogeography Analyses
2.4. Statistical Analysis
2.5. Data Accession Numbers
3. Results
3.1. Characteristics of the EBV+ Population
3.2. LMP1 Phylogenetic Analysis
3.3. Association of the Brazilian LMP1 Variants with Polymorphisms
3.4. Relationship of Brazilian LMP1 Variants with EBV Type and Zp Variants
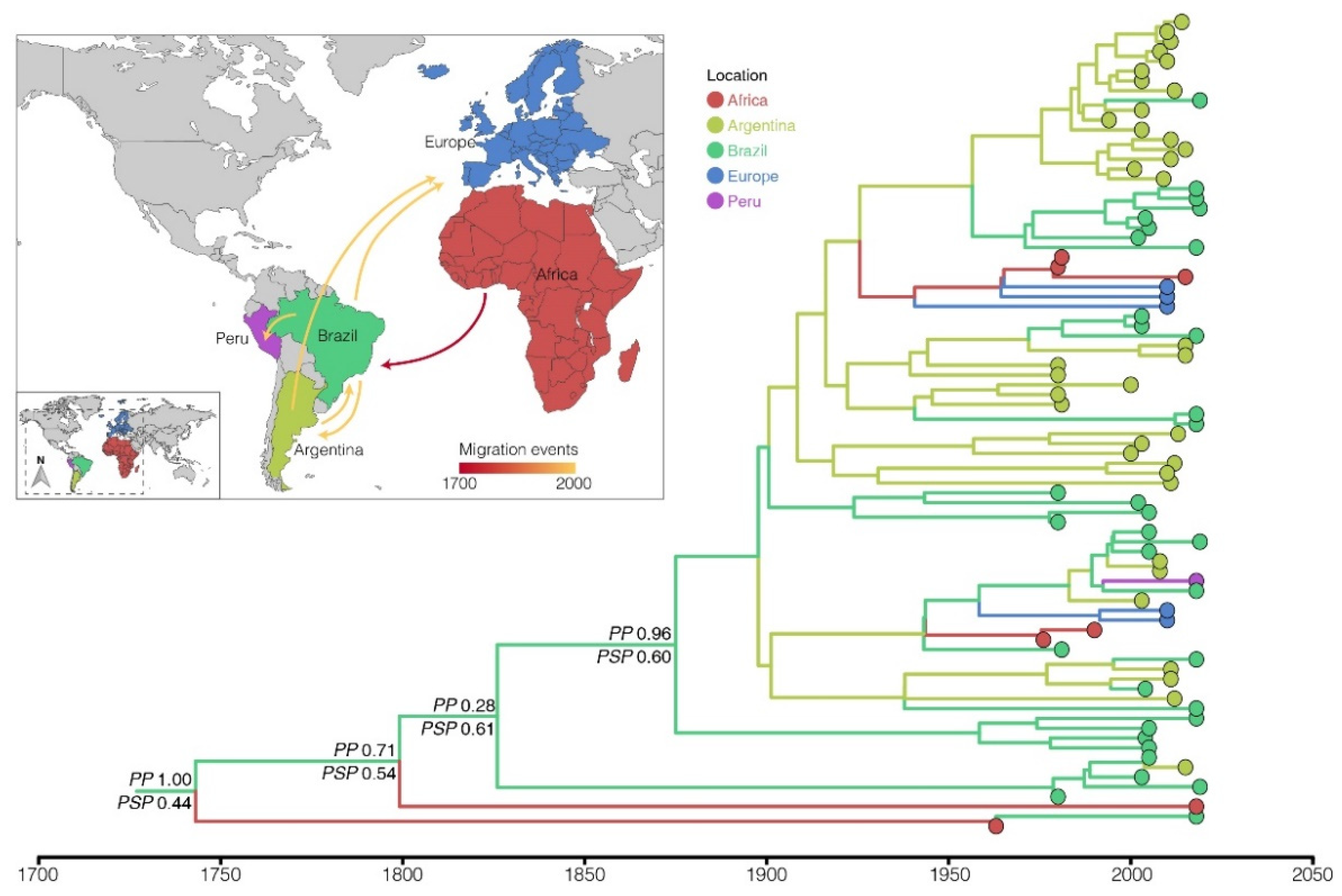
3.5. EBV Diversity and Spatial Dispersal in South America
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farrell, P.J. Epstein–Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Chabay, P.A.; Barros, M.H.M.; Hassan, R.; De Matteo, E.; Rey, G.; Carrico, M.K.; Renault, I.Z.; Preciado, M.V. Pediatric Hodgkin Lymphoma in 2 South American Series: A Distinctive Epidemiologic Pattern and Lack of Association of Epstein-Barr Virus with Clinical Outcome. J. Pediatr. Hematol. 2008, 30, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Klumb, C.E.; Felisbino, F.E.; Guiretti, D.M.; White, L.R.; Stefanoff, C.G.; Barros, M.H.M.; Seuánez, H.N.; Zalcberg, I.R. Clinical and Demographic Characteristics of Epstein-Barr Virus-Associated Childhood Burkitt’s Lymphoma in Southeastern Brazil: Epidemiological Insights from an Intermediate Risk Region. Haematologica 2008, 93, 780–783. [Google Scholar] [CrossRef]
- Barros, M.H.M.; Hassan, R.; Niedobitek, G. Disease Patterns in Pediatric Classical Hodgkin Lymphoma: A Report from a Developing Area in Brazil. Hematol. Oncol. 2011, 29, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Martín, P.; Krsnik, I.; Navarro, B.; Provencio, M.; García, J.F.; Bellas, C.; Vilches, C.; Gómez-Lozano, N. HLA Allele E*01:01 Is Associated with a Reduced Risk of EBV-Related Classical Hodgkin Lymphoma Independently of HLA-A*01/*02. PLoS ONE 2015, 10, e0135512. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. In Epstein Barr Virus Volume 1; Münz, C., Ed.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2015; Volume 390, pp. 151–209. ISBN 978-3-319-22821-1. [Google Scholar]
- Hadinoto, V.; Shapiro, M.; Greenough, T.C.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. On the Dynamics of Acute EBV Infection and the Pathogenesis of Infectious Mononucleosis. Blood 2008, 111, 1420–1427. [Google Scholar] [CrossRef]
- Tzellos, S.; Farrell, P.J. Epstein-Barr Virus Sequence Variation—Biology and Disease. Pathogens 2012, 1, 156–174. [Google Scholar] [CrossRef]
- Lei, H.; Li, T.; Li, B.; Tsai, S.; Biggar, R.J.; Nkrumah, F.; Neequaye, J.; Gutierrez, M.; Epelman, S.; Mbulaiteye, S.M.; et al. Epstein-Barr Virus from Burkitt Lymphoma Biopsies from Africa and South America Share Novel LMP-1 Promoter and Gene Variations. Sci. Rep. 2015, 5, 16706. [Google Scholar] [CrossRef]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A Cancer-Associated Epstein-Barr Virus BZLF1 Promoter Variant Enhances Lytic Infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef]
- Tong, J.H.M.; Lo, K.W.; Au, F.W.L.; Huang, D.P.; To, K.-F. Re: Discrete Alterations in the BZLF1 Promoter in Tumor and Non-Tumor-Associated Epstein-Barr Virus. JNCI J. Natl. Cancer Inst. 2003, 95, 1008–1009. [Google Scholar] [CrossRef]
- Martini, M.; Capello, D.; Serraino, D.; Navarra, A.; Pierconti, F.; Cenci, T.; Gaidano, G.; Larocca, L.M. Characterization of Variants in the Promoter of EBV Gene BZLF1 in Normal Donors, HIV-Positive Patients and in AIDS-Related Lymphomas. J. Infect. 2007, 54, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Bachmann, E.; Brousset, P.; Sandvej, K.; Nadal, D.; Bachmann, F.; Odermatt, B.; Delsol, G.; Pallesen, G. Deletions within the LMP1 Oncogene of Epstein-Barr Virus Are Clustered in Hodgkin’s Disease and Identical to Those Observed in Nasopharyngeal Carcinoma. Blood 1993, 82, 2937–2942. [Google Scholar] [CrossRef] [PubMed]
- Guiretti, D.M.; Chabay, P.A.; Valva, P.; Stefanoff, C.G.; Barros, M.H.M.; de Matteo, E.; Renault, I.Z.; Preciado, M.V.; Hassan, R. Structural Variability of the Carboxy-Terminus of Epstein–Barr Virus Encoded Latent Membrane Protein 1 Gene in Hodgkin’s Lymphomas. J. Med Virol. 2007, 79, 1722–1730. [Google Scholar] [CrossRef]
- Lorenzetti, M.A.; Gantuz, M.; Altcheh, J.; De Matteo, E.; Chabay, P.A.; Preciado, M.V. Distinctive Epstein-Barr Virus Variants Associated with Benign and Malignant Pediatric Pathologies: LMP1 Sequence Characterization and Linkage with Other Viral Gene Polymorphisms. J. Clin. Microbiol. 2012, 50, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba Abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome Diversity of Epstein-Barr Virus from Multiple Tumor Types and Normal Infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-M.; Liu, H.; Lei, H.; Li, B.; Chin, P.-J.; Tsai, S.; Bhatia, K.; Gutierrez, M.; Epelman, S.; Biggar, R.J.; et al. Frequency of EBV LMP-1 Promoter and Coding Variations in Burkitt Lymphoma Samples in Africa and South America and Peripheral Blood in Uganda. Cancers 2018, 10, 177. [Google Scholar] [CrossRef]
- Kaye, K.M.; Izumi, K.M.; Kieff, E. Epstein-Barr Virus Latent Membrane Protein 1 Is Essential for B-Lymphocyte Growth Transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 9150–9154. [Google Scholar] [CrossRef]
- Middeldorp, J.; Pegtel, D. Multiple roles of LMP1 in Epstein-Barr Virus Induced Immune Escape. Semin. Cancer Biol. 2008, 18, 388–396. [Google Scholar] [CrossRef]
- Lo, A.K.-F.; Dawson, C.W.; Lung, H.L.; Wong, K.-L.; Young, L.S. The Role of EBV-Encoded LMP1 in the NPC Tumor Microenvironment: From Function to Therapy. Front. Oncol. 2021, 11, 640207. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr Virus: Exploiting the Immune System. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Miller, W.E.; Edwards, R.H.; Walling, D.M.; Raab-Traub, N. Sequence Variation in the Epstein-Barr Virus Latent Membrane Protein 1. J. Gen. Virol. 1994, 75, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Berenstein, A.J.; Lorenzetti, M.A.; Preciado, M.V. Recombination Rates along the Entire Epstein Barr Virus Genome Display a Highly Heterogeneous Landscape. Infect. Genet. Evol. 2018, 65, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Fennewald, S.; van Santen, V.; Kieff, E. Nucleotide Sequence of an mRNA Transcribed in Latent Growth-Transforming Virus Infection Indicates That It May Encode a Membrane Protein. J. Virol. 1984, 51, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Van, D.; Enrberg, I.; Phi, P.P.-T.; Tran-Thi, C.; Hu, L. Epstein-Barr Virus Genetic Variation in Vietnamese Patients with Nasopharyngeal Carcinoma: Full-Length Analysis of LMP1. Virus Genes 2008, 37, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, E.; Butticaz, C.; Wyniger, J.; Martinez, R.; Battegay, M.; El Amari, E.B.; Dang, T.; Egger, J.-F.; Fehr, J.; Mueller-Garamvögyi, E.; et al. Genetic Diversity of EBV-Encoded LMP1 in the Swiss HIV Cohort Study and Implication for NF-Κb Activation. PLoS ONE 2012, 7, e32168. [Google Scholar] [CrossRef] [PubMed]
- Gantuz, M.; Lorenzetti, M.A.; Chabay, P.A.; Preciado, M.V. A Novel Recombinant Variant of Latent Membrane Protein 1 from Epstein Barr Virus in Argentina Denotes Phylogeographical Association. PLoS ONE 2017, 12, e0174221. [Google Scholar] [CrossRef]
- Walling, D.M.; Shebib, N.; Weaver, S.; Nichols, C.M.; Flaitz, C.M.; Webster-Cyriaque, J. The Molecular Epidemiology and Evolution of Epstein-Barr Virus: Sequence Variation and Genetic Recombination in the Latent Membrane Protein-1 Gene. J. Infect. Dis. 1999, 179, 763–774. [Google Scholar] [CrossRef]
- Edwards, R.H.; Seillier-Moiseiwitsch, F.; Raab-Traub, N. Signature Amino Acid Changes in Latent Membrane Protein 1 Distinguish Epstein–Barr Virus Strains. Virology 1999, 261, 79–95. [Google Scholar] [CrossRef]
- Sueur, C.; Lupo, J.; Mas, P.; Morand, P.; Boyer, V. Difference in Cytokine Production and Cell Cycle Progression Induced by Epstein-Barr Virus Lmp1 Deletion Variants in Kmh2, a Hodgkin Lymphoma Cell Line. Virol. J. 2014, 11, 94. [Google Scholar] [CrossRef]
- Correia, S.; Palser, A.; Karstegl, C.E.; Middeldorp, J.M.; Ramayanti, O.; Cohen, J.I.; Hildesheim, A.; Fellner, M.D.; Wiels, J.; White, R.E.; et al. Natural Variation of Epstein-Barr Virus Genes, Proteins, and Primary MicroRNA. J. Virol. 2017, 91, e00375-17. [Google Scholar] [CrossRef]
- Chang, C.M.; Yu, K.J.; Mbulaiteye, S.M.; Hildesheim, A.; Bhatia, K. The Extent of Genetic Diversity of Epstein-Barr Virus and Its Geographic and Disease Patterns: A Need for Reappraisal. Virus Res. 2009, 143, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Manzari, C.; Lionetti, C.; Mechelli, R.; Anastasiadou, E.; Buscarinu, M.C.; Ristori, G.; Salvetti, M.; Picardi, E.; D’Erchia, A.M.; et al. Geographic Population Structure in Epstein-Barr Virus Revealed by Comparative Genomics. Genome Biol. Evol. 2016, 8, 3284–3291. [Google Scholar] [CrossRef] [PubMed]
- McAulay, K.A.; Higgins, C.D.; Macsween, K.F.; Lake, A.; Jarrett, R.F.; Robertson, F.L.; Williams, H.; Crawford, D.H. HLA Class I Polymorphisms Are Associated with Development of Infectious Mononucleosis upon Primary EBV Infection. J. Clin. Investig. 2007, 117, 3042–3048. [Google Scholar] [CrossRef] [PubMed]
- Niens, M.; Jarrett, R.; Hepkema, B.; Nolte, I.M.; Diepstra, A.; Platteel, M.; Kouprie, N.; Delury, C.P.; Gallagher, A.; Visser, L.; et al. HLA-A*02 is Associated with a Reduced Risk and HLA-A*01 with an Increased Risk of Developing EBV+ Hodgkin Lymphoma. Blood 2007, 110, 3310–3315. [Google Scholar] [CrossRef] [PubMed]
- Karanikiotis, C.; Daniilidis, M.; Karyotis, N.; Bakogiannis, C.; Economopoulos, T.; Murray, S.; Papamichael, D.; Samantas, E.; Nikolaou, A.; Skoura, L.; et al. HLA Class II Alleles and the Presence of Circulating Epstein-Barr Virus DNA in Greek Patients with Nasopharyngeal Carcinoma. Strahlenther. Onkol. 2008, 184, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO Classification of Lymphoid Neoplasms and Beyond: Evolving Concepts and Practical Applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr Virus Types 1 and 2 Differ in their EBNA-3A, EBNA-3B, and EBNA-3C Genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef]
- Lorenzetti, M.A.; Gutiérrez, M.I.; Altcheh, J.; Moscatelli, G.; Moroni, S.; Chabay, P.A.; Preciado, M.V. Epstein-Barr Virus BZLF1 Gene Promoter Variants in Pediatric Patients with Acute Infectious Mononucleosis: Its Comparison with Pediatric Lymphomas. J. Med Virol. 2009, 81, 1912–1917. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Farrell, P.J.; White, R.E. Do Epstein–Barr Virus Mutations and Natural Genome Sequence Variations Contribute to Disease? Biomolecules 2021, 12, 17. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular Mechanisms of Viral Oncogenesis in Humans. Nat. Rev. Genet. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Hadhri-Guiga, B.; Khabir, A.-M.; Mokdad-Gargouri, R.; Ghorbel, A.-M.; Drira, M.; Daoud, J.; Frikha, M.; Jlidi, R.; Gargouri, A. Various 30 and 69bp Deletion Variants of the Epstein-Barr Virus LMP1 May Arise by Homologous Recombination in Nasopharyngeal Carcinoma of Tunisian Patients. Virus Res. 2006, 115, 24–30. [Google Scholar] [CrossRef]
- da Silva, Z.P.; de Almeida Ribeiro, M.C.S.; Barata, R.B.; de Almeida, M.F. Perfil Sociodemográfico e Padrão de Utilização dos Serviços de Saúde do Sistema Único de Saúde (SUS), 2003–2008. Ciênc. Saúde Coletiva 2011, 16, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Decaussin, G.; Leclerc, V.; Ooka, T. The Lytic Cycle of Epstein-Barr Virus in the Nonproducer Raji Line Can Be Rescued by The Expression of a 135-Kilodalton Protein Encoded by the BALF2 Open Reading Frame. J. Virol. 1995, 69, 7309–7314. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.; Bankier, A.T.; Barrell, B.G.; Farrell, P.J. Sequence Analysis of Raji Epstein-Barr Virus DNA. Virology 1988, 164, 334–340. [Google Scholar] [CrossRef]
- Badger, D.A.; Kuester, R.K.; Sauer, J.M.; Sipes, I.G. Gadolinium Chloride Reduces Cytochrome P450: Relevance to Chemical-Induced Hepatotoxicity. Toxicology. 1997, 121, 143–153. [Google Scholar] [CrossRef]
- Hassan, R.; White, L.R.; Stefanoff, C.G.; de Oliveira, D.E.; Felisbino, F.E.; Klumb, C.E.; Bacchi, C.E.; Seuánez, H.N.; Zalcberg, I.R. Epstein-Barr Virus (EBV) Detection and Typing by PCR: A Contribution to Diagnostic Screening of EBV-Positive Burkitt’s Lymphoma. Diagn. Pathol. 2006, 1, 17. [Google Scholar] [CrossRef]
- Monteiro, T.A.F.; Costa, I.B.; Costa, I.B.; dos Santos Corrêa, T.L.; Coelho, B.M.R.; Silva, A.E.S.; de Paula Ramos, F.L.; Filho, A.J.M.; Monteiro, J.L.F.; Siqueira, J.A.M.; et al. Genotypes of Epstein–Barr Virus (EBV1/EBV2) in Individuals with Infectious Mononucleosis in the Metropolitan Area of Belém, Brazil, between 2005 and 2016. Braz. J. Infect. Dis. 2020, 24, 322–329. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Gatherer, D. Lineage Structures in the Genome Sequences of Three Epstein–Barr Virus Strains. Virology 2007, 359, 1–5. [Google Scholar] [CrossRef]
- Zanella, L.; Riquelme, I.; Buchegger, K.; Abanto, M.; Ili, C.; Brebi, P. A Reliable Epstein-Barr Virus Classification Based on Phylogenomic and Population Analyses. Sci. Rep. 2019, 9, 9829. [Google Scholar] [CrossRef]
- Liao, H.-M.; Liu, H.; Chin, P.-J.; Li, B.; Hung, G.-C.; Tsai, S.; Otim, I.; Legason, I.D.; Ogwang, M.D.; Reynolds, S.J.; et al. Epstein-Barr Virus in Burkitt Lymphoma in Africa Reveals a Limited Set of Whole Genome and LMP-1 Sequence Patterns: Analysis of Archival Datasets and Field Samples from Uganda, Tanzania, and Kenya. Front. Oncol. 2022, 12, 812224. [Google Scholar] [CrossRef]
- Blazquez, A.C.; Berenstein, A.J.; Torres, C.; Izquierdo, A.; Lezama, C.; Moscatelli, G.; De Matteo, E.N.; Lorenzetti, M.A.; Preciado, M.V. Comprehensive Evolutionary Analysis of Complete Epstein–Barr Virus Genomes from Argentina and Other Geographies. Viruses 2021, 13, 1172. [Google Scholar] [CrossRef]
- Araujo, I.; Foss, H.; Bittencourt, A.; Hummel, M.; Demel, G.; Mendonca, N.; Herbst, H.; Stein, H. Expression of Epstein-Barr Virus-Gene Products in Burkitt’s Lymphoma in Northeast Brazil. Blood 1996, 87, 5279–5286. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, P.; Larrate, M.; Garcia-Costa, A.; Rohan, P.; Gama, B.E.; Abdelhay, E.; Delatorre, E.; Hassan, R. Spatial Dispersal of Epstein–Barr Virus in South America Reveals an African American Variant in Brazilian Lymphomas. Viruses 2022, 14, 1762. https://doi.org/10.3390/v14081762
Alves P, Larrate M, Garcia-Costa A, Rohan P, Gama BE, Abdelhay E, Delatorre E, Hassan R. Spatial Dispersal of Epstein–Barr Virus in South America Reveals an African American Variant in Brazilian Lymphomas. Viruses. 2022; 14(8):1762. https://doi.org/10.3390/v14081762
Chicago/Turabian StyleAlves, Paula, Marcella Larrate, Aruanã Garcia-Costa, Paulo Rohan, Bianca Ervatti Gama, Eliana Abdelhay, Edson Delatorre, and Rocio Hassan. 2022. "Spatial Dispersal of Epstein–Barr Virus in South America Reveals an African American Variant in Brazilian Lymphomas" Viruses 14, no. 8: 1762. https://doi.org/10.3390/v14081762