
Upstream of N-Ras (Unr/CSDE1) Interacts with NCp7 and Gag, Modulating HIV-1 IRES-Mediated Translation Initiation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture
2.2. Plasmids and Proteins
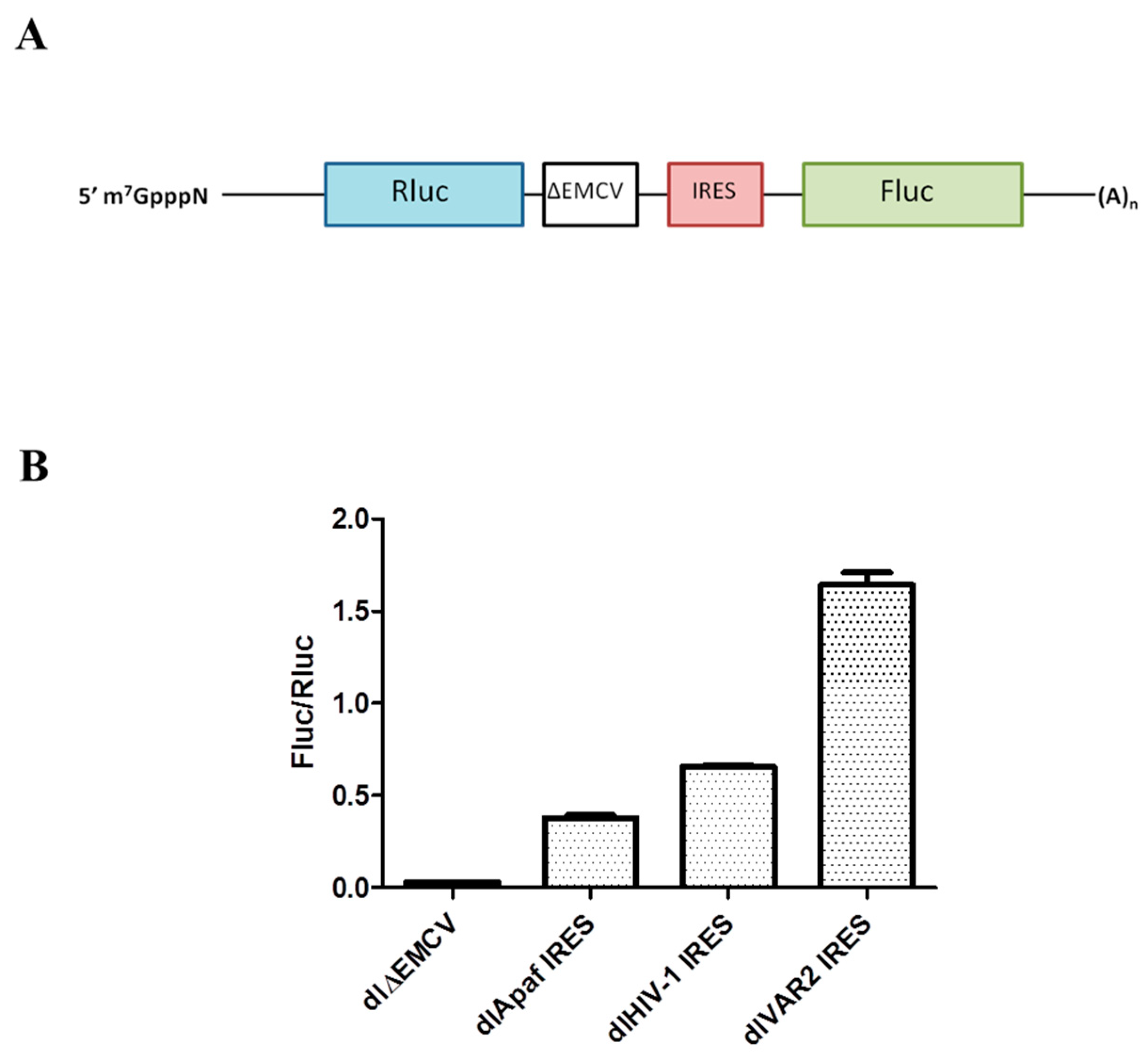
2.3. Dual Luciferase Assays
2.4. Co-Immunoprecipitation
2.5. Western Blots
2.6. Confocal Microscopy
2.7. Fluorescence Lifetime Imaging Microscopy (FLIM)
2.8. Viral Production and Quantification
2.9. Viral Infection
2.10. Quantitative PCR (qPCR)
3. Results
3.1. HIV-1 IRES Activity Is Stimulated by Unr
3.2. Identification of Unr Binding Site in the HIV-1 IRES
3.3. Opposite Effects of Gag and NCp7 on Unr-Promoted HIV-1 IRES Activity
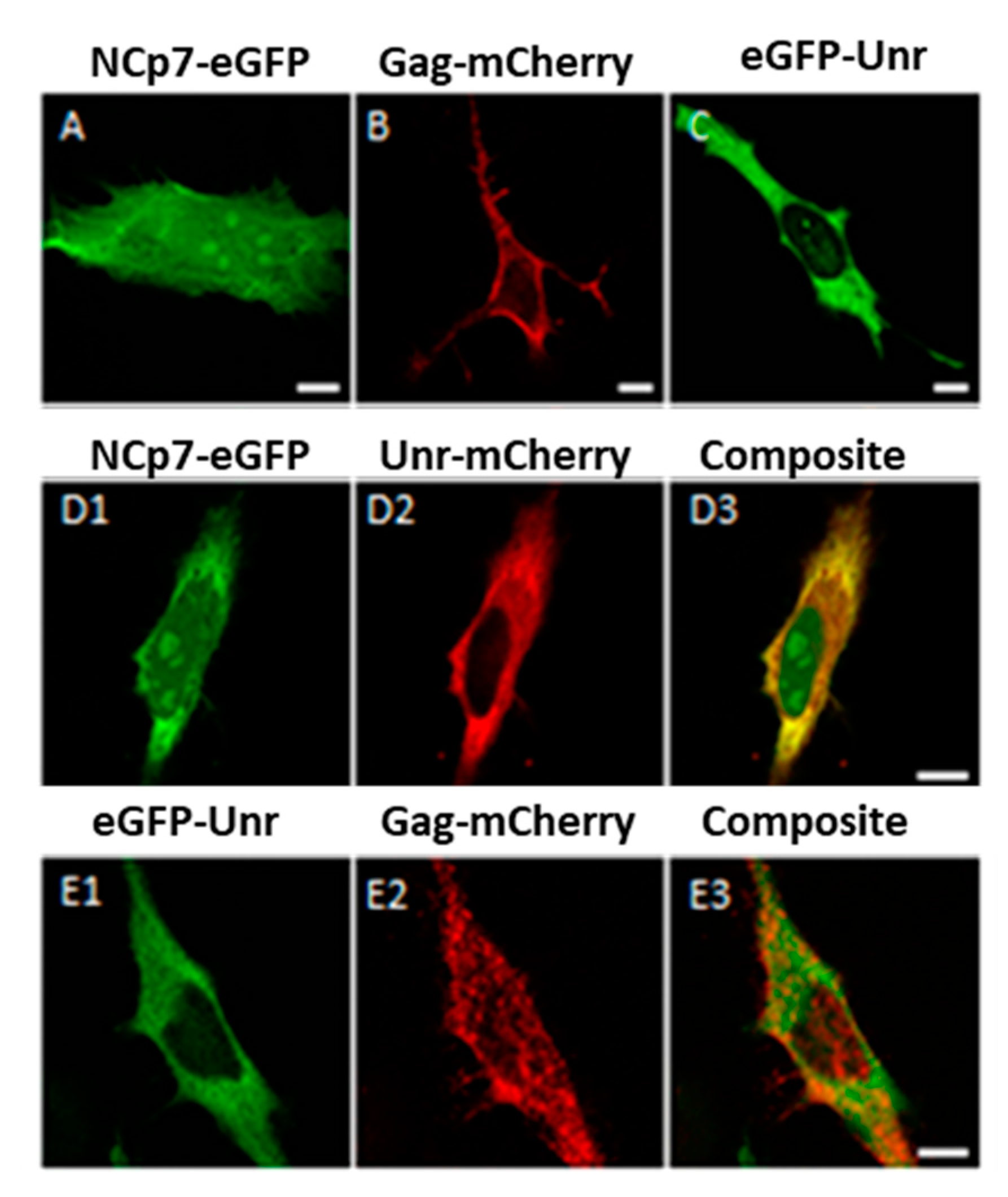
3.4. NCp7/Gag and Unr Co-Localize in the Cytoplasm
3.5. NCp7 Interaction with Unr Visualized by FRET-FLIM
3.6. Gag Interaction with Unr Visualized by FRET-FLIM
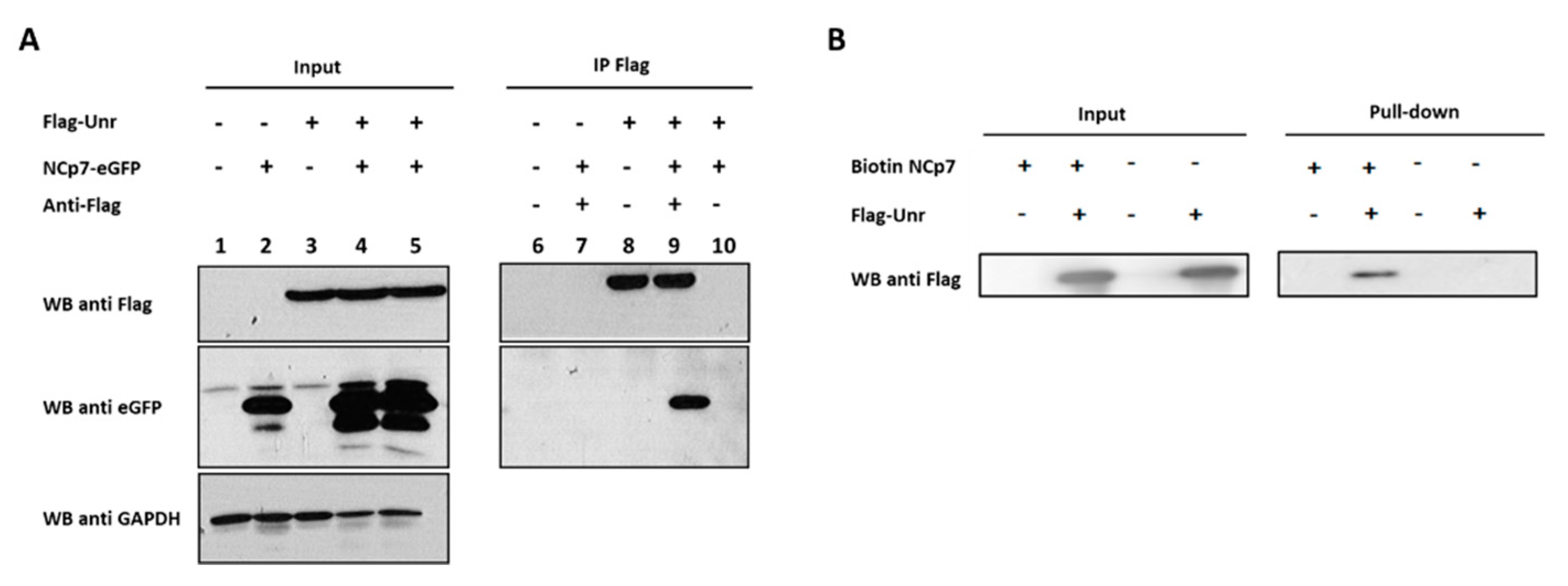
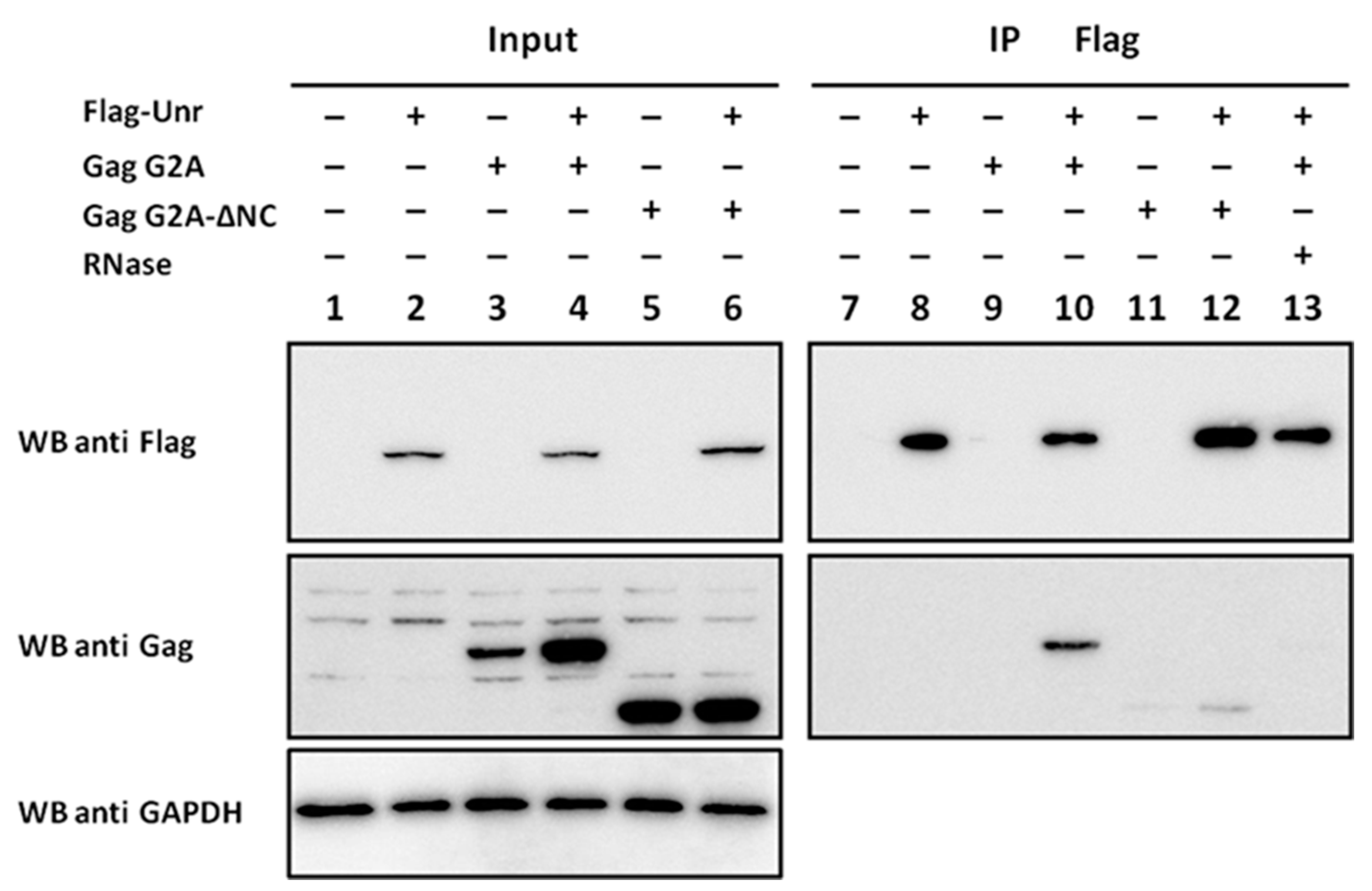
3.7. NCp7 and Gag Interaction with Unr Monitored by Co-Immunoprecipitation
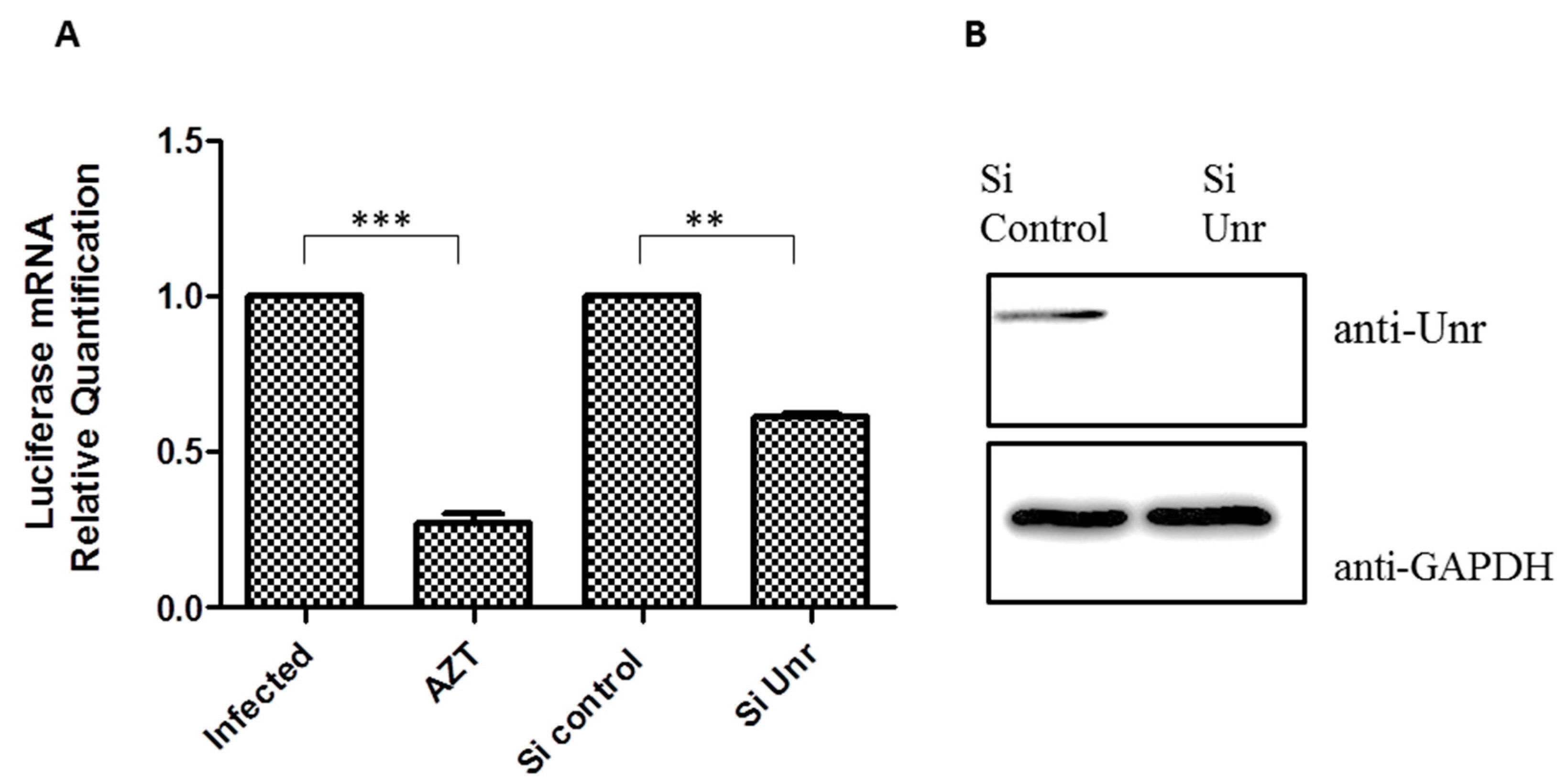
3.8. Unr Knockdown Reduces Infection by an HIV-1 Lentivector
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keane, S.C.; Heng, X.; Lu, K.; Kharytonchyk, S.; Ramakrishnan, V.; Carter, G.; Barton, S.; Hosic, A.; Florwick, A.; Santos, J.; et al. RNA Structure. Structure of the HIV-1 RNA Packaging Signal. Science 2015, 348, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Kharytonchyk, S.; Chaudry, I.; Iyer, A.S.; Carter, H.; Becker, G.; Desai, Y.; Glang, L.; Choi, S.H.; Singh, K.; et al. Structural Basis for Transcriptional Start Site Control of HIV-1 RNA Fate. Science 2020, 368, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Kharytonchyk, S.; Waller, A.; Mbaekwe, U.; Basappa, S.; Kuo, N.; Frank, H.M.; Quasney, C.; Kidane, A.; Swanson, C.; et al. Identification of the Initial Nucleocapsid Recognition Element in the HIV-1 RNA Packaging Signal. Proc. Natl. Acad. Sci. USA 2020, 117, 17737–17746. [Google Scholar] [CrossRef] [PubMed]
- Miele, G.; Mouland, A.; Harrison, G.P.; Cohen, E.; Lever, A.M. The Human Immunodeficiency Virus Type 1 5′ Packaging Signal Structure Affects Translation but Does Not Function as an Internal Ribosome Entry Site Structure. J. Virol. 1996, 70, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Arts, K.; Abbink, T.E.M. Ribosomal Scanning on the 5′-Untranslated Region of the Human Immunodeficiency Virus RNA Genome. Nucleic Acids Res. 2011, 39, 5232–5244. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.P.; Soto Rifo, R.; Herbreteau, C.H.; Decimo, D.; Ohlmann, T. Lentiviral RNAs Can Use Different Mechanisms for Translation Initiation. Biochem. Soc. Trans. 2008, 36, 690–693. [Google Scholar] [CrossRef]
- de Breyne, S.; Ohlmann, T. Focus on Translation Initiation of the HIV-1 MRNAs. Int. J. Mol. Sci. 2019, 20, 101. [Google Scholar] [CrossRef]
- Buck, C.B.; Shen, X.; Egan, M.A.; Pierson, T.C.; Walker, C.M.; Siliciano, R.F. The Human Immunodeficiency Virus Type 1 Gag Gene Encodes an Internal Ribosome Entry Site. J. Virol. 2001, 75, 181–191. [Google Scholar] [CrossRef]
- Herbreteau, C.H.; Weill, L.; Décimo, D.; Prévôt, D.; Darlix, J.-L.; Sargueil, B.; Ohlmann, T. HIV-2 Genomic RNA Contains a Novel Type of IRES Located Downstream of Its Initiation Codon. Nat. Struct. Mol. Biol. 2005, 12, 1001–1007. [Google Scholar] [CrossRef]
- Locker, N.; Chamond, N.; Sargueil, B. A Conserved Structure within the HIV Gag Open Reading Frame That Controls Translation Initiation Directly Recruits the 40S Subunit and EIF3. Nucleic Acids Res. 2011, 39, 2367–2377. [Google Scholar] [CrossRef]
- Weill, L.; James, L.; Ulryck, N.; Chamond, N.; Herbreteau, C.H.; Ohlmann, T.; Sargueil, B. A New Type of IRES within Gag Coding Region Recruits Three Initiation Complexes on HIV-2 Genomic RNA. Nucleic Acids Res. 2010, 38, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Brasey, A.; Lopez-Lastra, M.; Ohlmann, T.; Beerens, N.; Berkhout, B.; Darlix, J.-L.; Sonenberg, N. The Leader of Human Immunodeficiency Virus Type 1 Genomic RNA Harbors an Internal Ribosome Entry Segment That Is Active during the G2/M Phase of the Cell Cycle. J. Virol. 2003, 77, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Aravena, A.; Ramdohr, P.; Vallejos, M.; Valiente-Echeverría, F.; Dormoy-Raclet, V.; Rodríguez, F.; Pino, K.; Holzmann, C.; Huidobro-Toro, J.P.; Gallouzi, I.-E.; et al. The Elav-like Protein HuR Exerts Translational Control of Viral Internal Ribosome Entry Sites. Virology 2009, 392, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Vallejos, M.; Deforges, J.; Plank, T.-D.M.; Letelier, A.; Ramdohr, P.; Abraham, C.G.; Valiente-Echeverría, F.; Kieft, J.S.; Sargueil, B.; López-Lastra, M. Activity of the Human Immunodeficiency Virus Type 1 Cell Cycle-Dependent Internal Ribosomal Entry Site Is Modulated by IRES Trans-Acting Factors. Nucleic Acids Res. 2011, 39, 6186–6200. [Google Scholar] [CrossRef]
- Gendron, K.; Ferbeyre, G.; Heveker, N.; Brakier-Gingras, L. The Activity of the HIV-1 IRES Is Stimulated by Oxidative Stress and Controlled by a Negative Regulatory Element. Nucleic Acids Res. 2011, 39, 902–912. [Google Scholar] [CrossRef]
- Liu, J.; Henao-Mejia, J.; Liu, H.; Zhao, Y.; He, J.J. Translational Regulation of HIV-1 Replication by HIV-1 Rev Cellular Cofactors Sam68, EIF5A, HRIP, and DDX3. J. Neuroimmune Pharmacol. 2011, 6, 308–321. [Google Scholar] [CrossRef]
- Monette, A.; Ajamian, L.; López-Lastra, M.; Mouland, A.J. Human Immunodeficiency Virus Type 1 (HIV-1) Induces the Cytoplasmic Retention of Heterogeneous Nuclear Ribonucleoprotein A1 by Disrupting Nuclear Import: Implications for HIV-1 Gene Expression. J. Biol. Chem. 2009, 284, 31350–31362. [Google Scholar] [CrossRef]
- Vallejos, M.; Carvajal, F.; Pino, K.; Navarrete, C.; Ferres, M.; Huidobro-Toro, J.P.; Sargueil, B.; López-Lastra, M. Functional and Structural Analysis of the Internal Ribosome Entry Site Present in the MRNA of Natural Variants of the HIV-1. PLoS ONE 2012, 7, e35031. [Google Scholar] [CrossRef]
- Borman, A.; Howell, M.T.; Patton, J.G.; Jackson, R.J. The Involvement of a Spliceosome Component in Internal Initiation of Human Rhinovirus RNA Translation. J. Gen. Virol. 1993, 74 Pt 9, 1775–1788. [Google Scholar] [CrossRef]
- Meerovitch, K.; Svitkin, Y.V.; Lee, H.S.; Lejbkowicz, F.; Kenan, D.J.; Chan, E.K.; Agol, V.I.; Keene, J.D.; Sonenberg, N. La Autoantigen Enhances and Corrects Aberrant Translation of Poliovirus RNA in Reticulocyte Lysate. J. Virol. 1993, 67, 3798–3807. [Google Scholar] [CrossRef]
- Mitchell, S.A.; Brown, E.C.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. Protein Factor Requirements of the Apaf-1 Internal Ribosome Entry Segment: Roles of Polypyrimidine Tract Binding Protein and Upstream of N-Ras. Mol. Cell Biol. 2001, 21, 3364–3374. [Google Scholar] [CrossRef] [PubMed]
- Monette, A.; Valiente-Echeverría, F.; Rivero, M.; Cohen, É.A.; Lopez-Lastra, M.; Mouland, A.J. Dual Mechanisms of Translation Initiation of the Full-Length HIV-1 MRNA Contribute to Gag Synthesis. PLoS ONE 2013, 8, e68108. [Google Scholar] [CrossRef] [PubMed]
- Amorim, R.; Costa, S.M.; Cavaleiro, N.P.; da Silva, E.E.; da Costa, L.J. HIV-1 Transcripts Use IRES-Initiation under Conditions Where Cap-Dependent Translation Is Restricted by Poliovirus 2A Protease. PLoS ONE 2014, 9, e88619. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.E.; Wu, L.I.; Emerman, M. The Human Immunodeficiency Virus Type 1 Vpr Gene Prevents Cell Proliferation during Chronic Infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [CrossRef] [PubMed]
- King, H.A.; Cobbold, L.C.; Willis, A.E. The Role of IRES Trans-Acting Factors in Regulating Translation Initiation. Biochem. Soc. Trans. 2010, 38, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Balvay, L.; Soto Rifo, R.; Ricci, E.P.; Decimo, D.; Ohlmann, T. Structural and Functional Diversity of Viral IRESes. Biochim. Biophys. Acta 2009, 1789, 542–557. [Google Scholar] [CrossRef]
- Filbin, M.E.; Kieft, J.S. Toward a Structural Understanding of IRES RNA Function. Curr. Opin. Struct. Biol. 2009, 19, 267–276. [Google Scholar] [CrossRef]
- Fitzgerald, K.D.; Semler, B.L. Bridging IRES Elements in MRNAs to the Eukaryotic Translation Apparatus. Biochim. Biophys. Acta 2009, 1789, 518–528. [Google Scholar] [CrossRef]
- Jacquemin-Sablon, H.; Triqueneaux, G.; Deschamps, S.; le Maire, M.; Doniger, J.; Dautry, F. Nucleic Acid Binding and Intracellular Localization of Unr, a Protein with Five Cold Shock Domains. Nucleic Acids Res. 1994, 22, 2643–2650. [Google Scholar] [CrossRef]
- Ferrer, N.; Garcia-Espana, A.; Jeffers, M.; Pellicer, A. The Unr Gene: Evolutionary Considerations and Nucleic Acid-Binding Properties of Its Long Isoform Product. DNA Cell Biol. 1999, 18, 209–218. [Google Scholar] [CrossRef]
- Saltel, F.; Giese, A.; Azzi, L.; Elatmani, H.; Costet, P.; Ezzoukhry, Z.; Dugot-Senant, N.; Miquerol, L.; Boussadia, O.; Wodrich, H.; et al. Unr Defines a Novel Class of Nucleoplasmic Reticulum Involved in MRNA Translation. J. Cell Sci. 2017, 130, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, N.M.; Jagtap, P.K.A.; Masiewicz, P.; Guitart, T.; Simon, B.; Provaznik, J.; Stein, F.; Haberkant, P.; Sweetapple, L.J.; Villacorta, L.; et al. Pseudo-RNA-Binding Domains Mediate RNA Structure Specificity in Upstream of N-Ras. Cell Rep. 2020, 32, 107930. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, O.; Niepmann, M.; Créancier, L.; Prats, A.-C.; Dautry, F.; Jacquemin-Sablon, H. Unr Is Required in Vivo for Efficient Initiation of Translation from the Internal Ribosome Entry Sites of Both Rhinovirus and Poliovirus. J. Virol. 2003, 77, 3353–3359. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.C.; Jackson, R.J. All Five Cold-Shock Domains of Unr (Upstream of N-Ras) Are Required for Stimulation of Human Rhinovirus RNA Translation. J. Gen. Virol. 2004, 85, 2279–2287. [Google Scholar] [CrossRef]
- Mitchell, S.A.; Spriggs, K.A.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. The Apaf-1 Internal Ribosome Entry Segment Attains the Correct Structural Conformation for Function via Interactions with PTB and Unr. Mol. Cell 2003, 11, 757–771. [Google Scholar] [CrossRef]
- Tinton, S.A.; Schepens, B.; Bruynooghe, Y.; Beyaert, R.; Cornelis, S. Regulation of the Cell-Cycle-Dependent Internal Ribosome Entry Site of the PITSLRE Protein Kinase: Roles of Unr (Upstream of N-Ras) Protein and Phosphorylated Translation Initiation Factor EIF-2alpha. Biochem. J. 2005, 385, 155–163. [Google Scholar] [CrossRef]
- Clever, J.; Sassetti, C.; Parslow, T.G. RNA Secondary Structure and Binding Sites for Gag Gene Products in the 5′ Packaging Signal of Human Immunodeficiency Virus Type 1. J. Virol. 1995, 69, 2101–2109. [Google Scholar] [CrossRef]
- Clever, J.L.; Miranda, D.; Parslow, T.G. RNA Structure and Packaging Signals in the 5′ Leader Region of the Human Immunodeficiency Virus Type 1 Genome. J. Virol. 2002, 76, 12381–12387. [Google Scholar] [CrossRef]
- Dannull, J.; Surovoy, A.; Jung, G.; Moelling, K. Specific Binding of HIV-1 Nucleocapsid Protein to PSI RNA in Vitro Requires N-Terminal Zinc Finger and Flanking Basic Amino Acid Residues. EMBO J. 1994, 13, 1525–1533. [Google Scholar] [CrossRef]
- Darlix, J.-L.; Garrido, J.L.; Morellet, N.; Mély, Y.; de Rocquigny, H. Properties, Functions, and Drug Targeting of the Multifunctional Nucleocapsid Protein of the Human Immunodeficiency Virus. Adv. Pharmacol. 2007, 55, 299–346. [Google Scholar] [CrossRef]
- Darlix, J.L.; Lapadat-Tapolsky, M.; de Rocquigny, H.; Roques, B.P. First Glimpses at Structure-Function Relationships of the Nucleocapsid Protein of Retroviruses. J. Mol. Biol. 1995, 254, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Gorelick, R.J. Nucleocapsid Protein Function in Early Infection Processes. Virus Res. 2008, 134, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Darlix, J.-L.; Godet, J.; Ivanyi-Nagy, R.; Fossé, P.; Mauffret, O.; Mély, Y. Flexible Nature and Specific Functions of the HIV-1 Nucleocapsid Protein. J. Mol. Biol. 2011, 410, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, R.; Fisher, J.; Goff, S.P. RNA Packaging. Curr. Top. Microbiol. Immunol. 1996, 214, 177–218. [Google Scholar] [CrossRef] [PubMed]
- Sakuragi, J.; Shioda, T.; Panganiban, A.T. Duplication of the Primary Encapsidation and Dimer Linkage Region of Human Immunodeficiency Virus Type 1 RNA Results in the Appearance of Monomeric RNA in Virions. J. Virol. 2001, 75, 2557–2565. [Google Scholar] [CrossRef]
- Ding, P.; Kharytonchyk, S.; Kuo, N.; Cannistraci, E.; Flores, H.; Chaudhary, R.; Sarkar, M.; Dong, X.; Telesnitsky, A.; Summers, M.F. 5′-Cap Sequestration Is an Essential Determinant of HIV-1 Genome Packaging. Proc. Natl. Acad. Sci. USA 2021, 118, e2112475118. [Google Scholar] [CrossRef]
- Berkhout, B.; van Wamel, J.L. Role of the DIS Hairpin in Replication of Human Immunodeficiency Virus Type 1. J. Virol. 1996, 70, 6723–6732. [Google Scholar] [CrossRef]
- Clever, J.L.; Parslow, T.G. Mutant Human Immunodeficiency Virus Type 1 Genomes with Defects in RNA Dimerization or Encapsidation. J. Virol. 1997, 71, 3407–3414. [Google Scholar] [CrossRef]
- El Meshri, S.E.; Dujardin, D.; Godet, J.; Richert, L.; Boudier, C.; Darlix, J.L.; Didier, P.; Mély, Y.; de Rocquigny, H. Role of the Nucleocapsid Domain in HIV-1 Gag Oligomerization and Trafficking to the Plasma Membrane: A Fluorescence Lifetime Imaging Microscopy Investigation. J. Mol. Biol. 2015, 427, 1480–1494. [Google Scholar] [CrossRef]
- de Rocquigny, H.; El Meshri, S.E.; Richert, L.; Didier, P.; Darlix, J.-L.; Mély, Y. Role of the Nucleocapsid Region in HIV-1 Gag Assembly as Investigated by Quantitative Fluorescence-Based Microscopy. Virus Res. 2014, 193, 78–88. [Google Scholar] [CrossRef]
- Cimarelli, A.; Sandin, S.; Höglund, S.; Luban, J. Basic Residues in Human Immunodeficiency Virus Type 1 Nucleocapsid Promote Virion Assembly via Interaction with RNA. J. Virol. 2000, 74, 3046–3057. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, T.; Luban, J.; Goff, S.P.; Haseltine, W.A.; Göttlinger, H.G. Mapping of Functionally Important Residues of a Cysteine-Histidine Box in the Human Immunodeficiency Virus Type 1 Nucleocapsid Protein. J. Virol. 1993, 67, 6159–6169. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, R.J.; Nigida, S.M.; Bess, J.W.; Arthur, L.O.; Henderson, L.E.; Rein, A. Noninfectious Human Immunodeficiency Virus Type 1 Mutants Deficient in Genomic RNA. J. Virol. 1990, 64, 3207–3211. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global Landscape of HIV-Human Protein Complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Boutant, E.; Bonzi, J.; Anton, H.; Nasim, M.B.; Cathagne, R.; Réal, E.; Dujardin, D.; Carl, P.; Didier, P.; Paillart, J.-C.; et al. Zinc Fingers in HIV-1 Gag Precursor Are Not Equivalent for GRNA Recruitment at the Plasma Membrane. Biophys. J. 2020, 119, 419–433. [Google Scholar] [CrossRef] [PubMed]
- De Rocquigny, H.; Gabus, C.; Vincent, A.; Fournié-Zaluski, M.C.; Roques, B.; Darlix, J.L. Viral RNA Annealing Activities of Human Immunodeficiency Virus Type 1 Nucleocapsid Protein Require Only Peptide Domains Outside the Zinc Fingers. Proc. Natl. Acad. Sci. USA 1992, 89, 6472–6476. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.H.; Fouchier, R.A.; Southerling, T.E.; Guerra, C.B.; Grant, C.K.; Malim, M.H. The Vif and Gag Proteins of Human Immunodeficiency Virus Type 1 Colocalize in Infected Human T Cells. J. Virol. 1997, 71, 5259–5267. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Meyer, B.E.; Simon, J.H.; Fischer, U.; Malim, M.H. HIV-1 Infection of Non-Dividing Cells: Evidence That the Amino-Terminal Basic Region of the Viral Matrix Protein Is Important for Gag Processing but Not for Post-Entry Nuclear Import. EMBO J. 1997, 16, 4531–4539. [Google Scholar] [CrossRef]
- Azoulay, J.; Clamme, J.P.; Darlix, J.L.; Roques, B.P.; Mély, Y. Destabilization of the HIV-1 Complementary Sequence of TAR by the Nucleocapsid Protein through Activation of Conformational Fluctuations. J. Mol. Biol. 2003, 326, 691–700. [Google Scholar] [CrossRef]
- Clamme, J.-P.; Krishnamoorthy, G.; Mély, Y. Intracellular Dynamics of the Gene Delivery Vehicle Polyethylenimine during Transfection: Investigation by Two-Photon Fluorescence Correlation Spectroscopy. Biochim. Biophys. Acta 2003, 1617, 52–61. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Echeverría, F.; Vallejos, M.; Monette, A.; Pino, K.; Letelier, A.; Huidobro-Toro, J.P.; Mouland, A.J.; López-Lastra, M. A Cis-Acting Element Present within the Gag Open Reading Frame Negatively Impacts on the Activity of the HIV-1 IRES. PLoS ONE 2013, 8, e56962. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B. Structure and Function of the Human Immunodeficiency Virus Leader RNA. Prog. Nucleic Acid. Res. Mol. Biol. 1996, 54, 1–34. [Google Scholar] [CrossRef]
- Coldwell, M.J.; Mitchell, S.A.; Stoneley, M.; MacFarlane, M.; Willis, A.E. Initiation of Apaf-1 Translation by Internal Ribosome Entry. Oncogene 2000, 19, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Triqueneaux, G.; Velten, M.; Franzon, P.; Dautry, F.; Jacquemin-Sablon, H. RNA Binding Specificity of Unr, a Protein with Five Cold Shock Domains. Nucleic Acids Res. 1999, 27, 1926–1934. [Google Scholar] [CrossRef]
- Plank, T.-D.M.; Whitehurst, J.T.; Kieft, J.S. Cell Type Specificity and Structural Determinants of IRES Activity from the 5′ Leaders of Different HIV-1 Transcripts. Nucleic Acids Res. 2013, 41, 6698–6714. [Google Scholar] [CrossRef]
- Abd El-Wahab, E.W.; Smyth, R.P.; Mailler, E.; Bernacchi, S.; Vivet-Boudou, V.; Hijnen, M.; Jossinet, F.; Mak, J.; Paillart, J.-C.; Marquet, R. Specific Recognition of the HIV-1 Genomic RNA by the Gag Precursor. Nat. Commun. 2014, 5, 4304. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Gorelick, R.J.; Vasa, S.M.; Guex, N.; Rein, A.; Mathews, D.H.; Giddings, M.C.; Weeks, K.M. High-Throughput SHAPE Analysis Reveals Structures in HIV-1 Genomic RNA Strongly Conserved across Distinct Biological States. PLoS Biol. 2008, 6, e96. [Google Scholar] [CrossRef]
- Wu, T.; Datta, S.A.K.; Mitra, M.; Gorelick, R.J.; Rein, A.; Levin, J.G. Fundamental Differences between the Nucleic Acid Chaperone Activities of HIV-1 Nucleocapsid Protein and Gag or Gag-Derived Proteins: Biological Implications. Virology 2010, 405, 556–567. [Google Scholar] [CrossRef]
- Cen, S.; Khorchid, A.; Gabor, J.; Rong, L.; Wainberg, M.A.; Kleiman, L. Roles of Pr55(Gag) and NCp7 in TRNA(3)(Lys) Genomic Placement and the Initiation Step of Reverse Transcription in Human Immunodeficiency Virus Type 1. J. Virol. 2000, 74, 10796–10800. [Google Scholar] [CrossRef]
- Cruceanu, M.; Urbaneja, M.A.; Hixson, C.V.; Johnson, D.G.; Datta, S.A.; Fivash, M.J.; Stephen, A.G.; Fisher, R.J.; Gorelick, R.J.; Casas-Finet, J.R.; et al. Nucleic Acid Binding and Chaperone Properties of HIV-1 Gag and Nucleocapsid Proteins. Nucleic Acids Res. 2006, 34, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Karnib, H.; Nadeem, M.F.; Humbert, N.; Sharma, K.K.; Grytsyk, N.; Tisné, C.; Boutant, E.; Lequeu, T.; Réal, E.; Boudier, C.; et al. The Nucleic Acid Chaperone Activity of the HIV-1 Gag Polyprotein Is Boosted by Its Cellular Partner RPL7: A Kinetic Study. Nucleic Acids Res. 2020, 48, 9218–9234. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.C.; Demarco, I.A.; Day, R.N. Quantitative Imaging of Protein Interactions in the Cell Nucleus. Biotechniques 2005, 38, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Day, R.N.; Periasamy, A.; Schaufele, F. Fluorescence Resonance Energy Transfer Microscopy of Localized Protein Interactions in the Living Cell Nucleus. Methods 2001, 25, 4–18. [Google Scholar] [CrossRef]
- Bastiaens, P.I.; Squire, A. Fluorescence Lifetime Imaging Microscopy: Spatial Resolution of Biochemical Processes in the Cell. Trends Cell Biol. 1999, 9, 48–52. [Google Scholar] [CrossRef]
- Barrera, A.; Ramos, H.; Vera-Otarola, J.; Fernández-García, L.; Angulo, J.; Olguín, V.; Pino, K.; Mouland, A.J.; López-Lastra, M. Post-Translational Modifications of HnRNP A1 Differentially Modulate Retroviral IRES-Mediated Translation Initiation. Nucleic Acids Res. 2020, 48, 10479–10499. [Google Scholar] [CrossRef]
- Ramos, H.; Monette, A.; Niu, M.; Barrera, A.; López-Ulloa, B.; Fuentes, Y.; Guizar, P.; Pino, K.; DesGroseillers, L.; Mouland, A.J.; et al. The Double-Stranded RNA-Binding Protein, Staufen1, Is an IRES-Transacting Factor Regulating HIV-1 Cap-Independent Translation Initiation. Nucleic Acids Res. 2022, 50, 411–429. [Google Scholar] [CrossRef]
- Carvajal, F.; Vallejos, M.; Walters, B.; Contreras, N.; Hertz, M.I.; Olivares, E.; Cáceres, C.J.; Pino, K.; Letelier, A.; Thompson, S.R.; et al. Structural Domains within the HIV-1 MRNA and the Ribosomal Protein S25 Influence Cap-Independent Translation Initiation. FEBS J. 2016, 283, 2508–2527. [Google Scholar] [CrossRef]
- Levin, J.G.; Mitra, M.; Mascarenhas, A.; Musier-Forsyth, K. Role of HIV-1 Nucleocapsid Protein in HIV-1 Reverse Transcription. RNA Biol. 2010, 7, 754–774. [Google Scholar] [CrossRef]
- Anderson, E.C.; Lever, A.M.L. Human Immunodeficiency Virus Type 1 Gag Polyprotein Modulates Its Own Translation. J. Virol. 2006, 80, 10478–10486. [Google Scholar] [CrossRef]
- Roldan, A.; Warren, O.U.; Russell, R.S.; Liang, C.; Wainberg, M.A. A HIV-1 Minimal Gag Protein Is Superior to Nucleocapsid at in Vitro Annealing and Exhibits Multimerization-Induced Inhibition of Reverse Transcription. J. Biol. Chem. 2005, 280, 17488–17496. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.P.; Datta, S.A.K.; Rein, A.; Rouzina, I.; Musier-Forsyth, K. Matrix Domain Modulates HIV-1 Gag’s Nucleic Acid Chaperone Activity via Inositol Phosphate Binding. J. Virol. 2011, 85, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.L.; Hsuan, J.J.; Totty, N.; Jackson, R.J. Unr, a Cellular Cytoplasmic RNA-Binding Protein with Five Cold-Shock Domains, Is Required for Internal Initiation of Translation of Human Rhinovirus RNA. Genes Dev. 1999, 13, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Athavale, S.S.; Ouyang, W.; McPike, M.P.; Hudson, B.S.; Borer, P.N. Effects of the Nature and Concentration of Salt on the Interaction of the HIV-1 Nucleocapsid Protein with SL3 RNA. Biochemistry 2010, 49, 3525–3533. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.J.; Rein, A.; Fivash, M.; Urbaneja, M.A.; Casas-Finet, J.R.; Medaglia, M.; Henderson, L.E. Sequence-Specific Binding of Human Immunodeficiency Virus Type 1 Nucleocapsid Protein to Short Oligonucleotides. J. Virol. 1998, 72, 1902–1909. [Google Scholar] [CrossRef]
- Burnett, A.; Spearman, P. APOBEC3G Multimers Are Recruited to the Plasma Membrane for Packaging into Human Immunodeficiency Virus Type 1 Virus-like Particles in an RNA-Dependent Process Requiring the NC Basic Linker. J. Virol. 2007, 81, 5000–5013. [Google Scholar] [CrossRef]
- Zhao, R.Y.; Li, G.; Bukrinsky, M.I. Vpr-Host Interactions during HIV-1 Viral Life Cycle. J. Neuroimmune Pharmacol. 2011, 6, 216–229. [Google Scholar] [CrossRef]
- Webb, J.A.; Jones, C.P.; Parent, L.J.; Rouzina, I.; Musier-Forsyth, K. Distinct Binding Interactions of HIV-1 Gag to Psi and Non-Psi RNAs: Implications for Viral Genomic RNA Packaging. RNA 2013, 19, 1078–1088. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IRES mut rev | (Phos)TGTTCGGGCGCCACTGCTAGAG |
| IRES mut U fwd | (Phos)GGGACACTTGAAAGCGATTCATAAGCCAGAGGAG |
| IRES mut C fwd | (Phos)GGGACTTGAAAGCGACCAGCAAGCCAGAGGAG |
| IRES mut 211 fwd | (Phos)GGGACTTGAAAGCGAAGGTAAAGCCAGAGGAG |
| IRES mut 212 fwd | (Phos)GGGACTTGAAAGCGAAACTAAAGCCAGAGGAG |
| IRES mut 213 fwd | (Phos)GGGACTTGAAAGCGAAAGAAAAGCCAGAGGAG |
| IRES mut 214 fwd | (Phos)GGGACTTGAAAGCGAAAGTGAAGCCAGAGGAG |
| Clone | Sequence |
|---|---|
| Wild-type sequence | 210 AAGUA 214 |
| mutU | 210 UUCAU 214 |
| mutC | 210 CCAGC 214 |
| mut211 | 210 AGGUA 214 |
| mut212 | 210 AACUA 214 |
| mut213 | 210 AAGAA 214 |
| mut214 | 210 AAGUG 214 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taha, N.; Zgheib, S.; Sharma, K.K.; Humbert, N.; Boutant, E.; Didier, P.; Mély, Y.; Real, E. Upstream of N-Ras (Unr/CSDE1) Interacts with NCp7 and Gag, Modulating HIV-1 IRES-Mediated Translation Initiation. Viruses 2022, 14, 1798. https://doi.org/10.3390/v14081798
Taha N, Zgheib S, Sharma KK, Humbert N, Boutant E, Didier P, Mély Y, Real E. Upstream of N-Ras (Unr/CSDE1) Interacts with NCp7 and Gag, Modulating HIV-1 IRES-Mediated Translation Initiation. Viruses. 2022; 14(8):1798. https://doi.org/10.3390/v14081798
Chicago/Turabian StyleTaha, Nedal, Sarwat Zgheib, Kamal Kant Sharma, Nicolas Humbert, Emmanuel Boutant, Pascal Didier, Yves Mély, and Eleonore Real. 2022. "Upstream of N-Ras (Unr/CSDE1) Interacts with NCp7 and Gag, Modulating HIV-1 IRES-Mediated Translation Initiation" Viruses 14, no. 8: 1798. https://doi.org/10.3390/v14081798