
Scratching the Surface Takes a Toll: Immune Recognition of Viral Proteins by Surface Toll-like Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
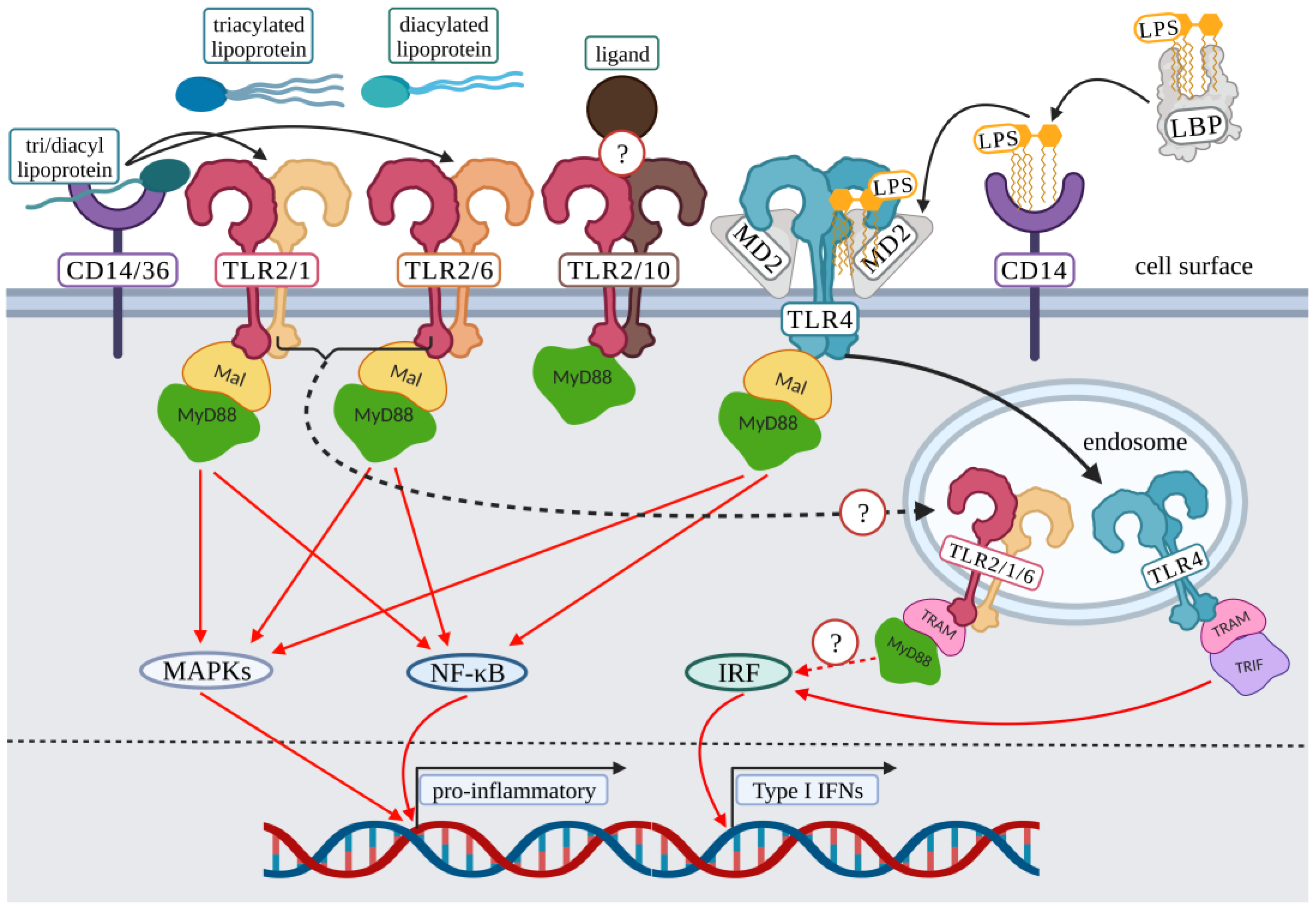
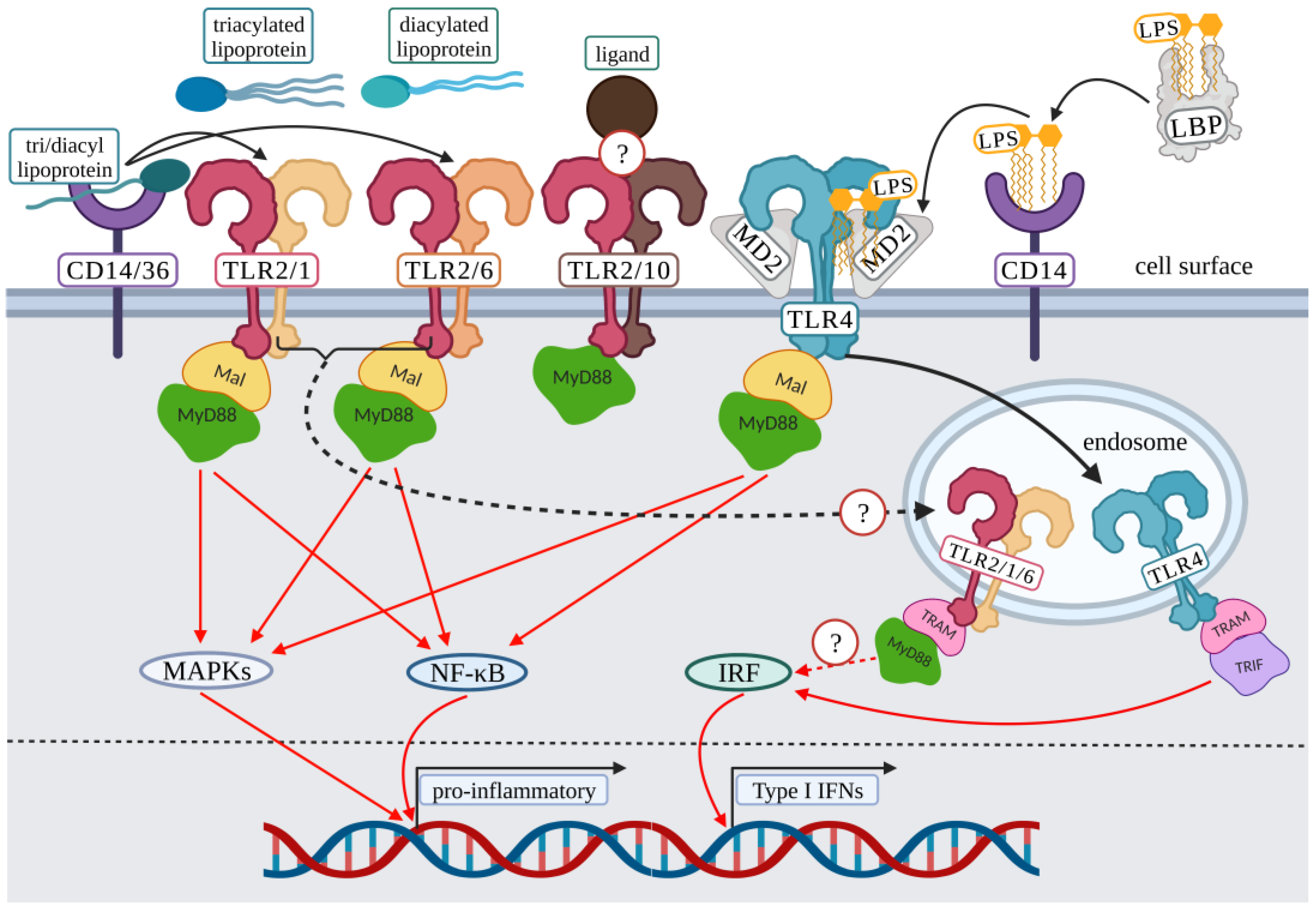
2. Surface Toll-Like Receptor Signaling: Initiation
3. Surface TLR Associated Virus Interactions
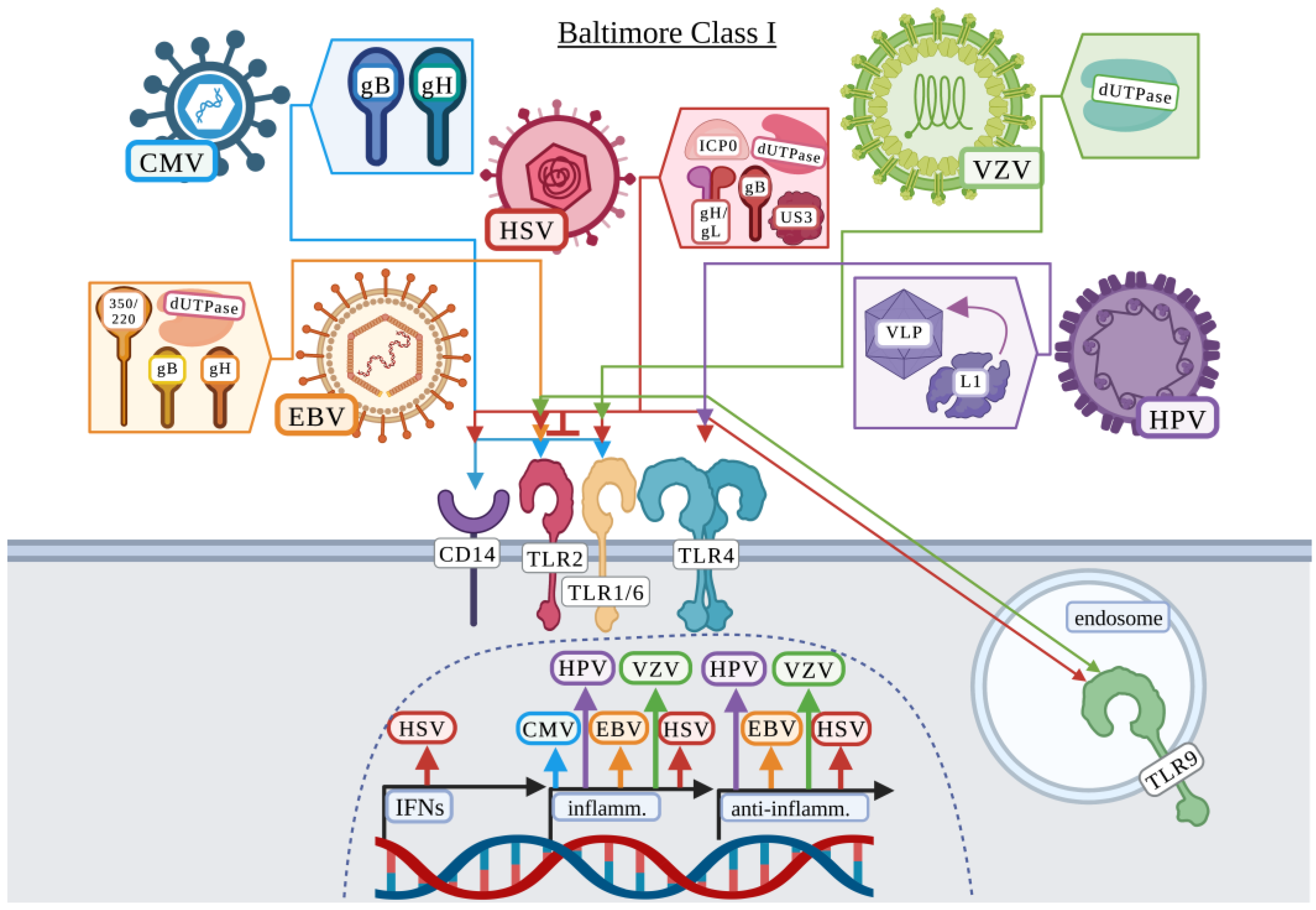
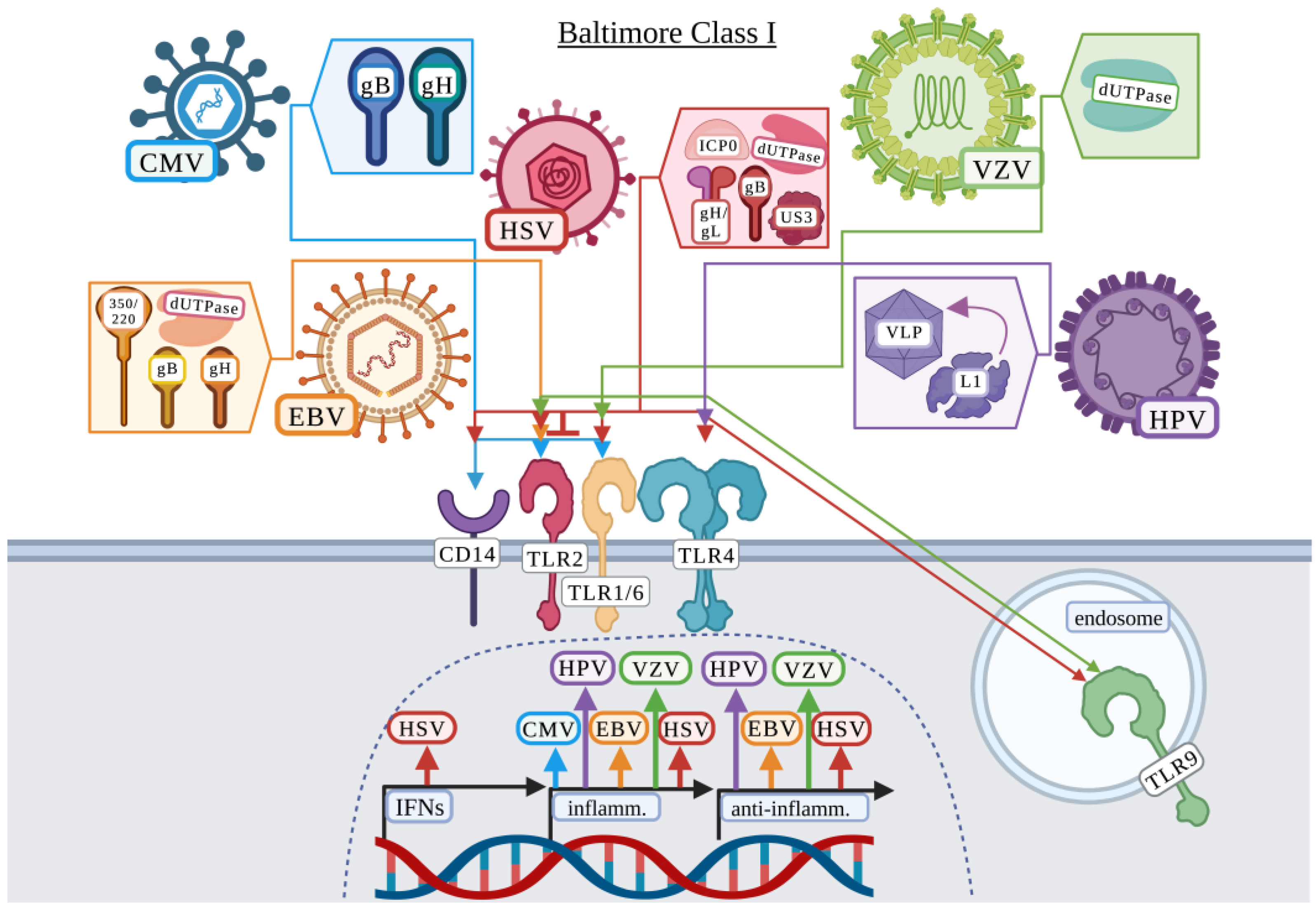
3.1. Baltimore Classification: Class I
3.1.1. Cytomegalovirus
3.1.2. Epstein–Barr Virus
3.1.3. Herpes Simplex Virus
3.1.4. Human Papillomavirus
3.1.5. Varicella-Zoster Virus
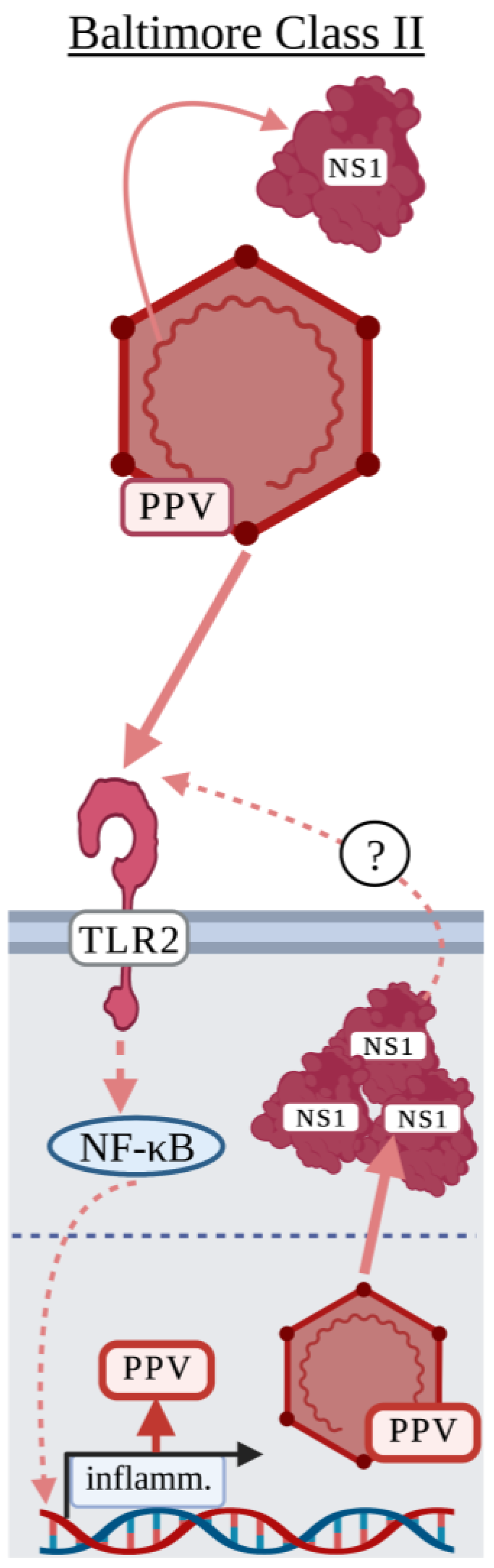
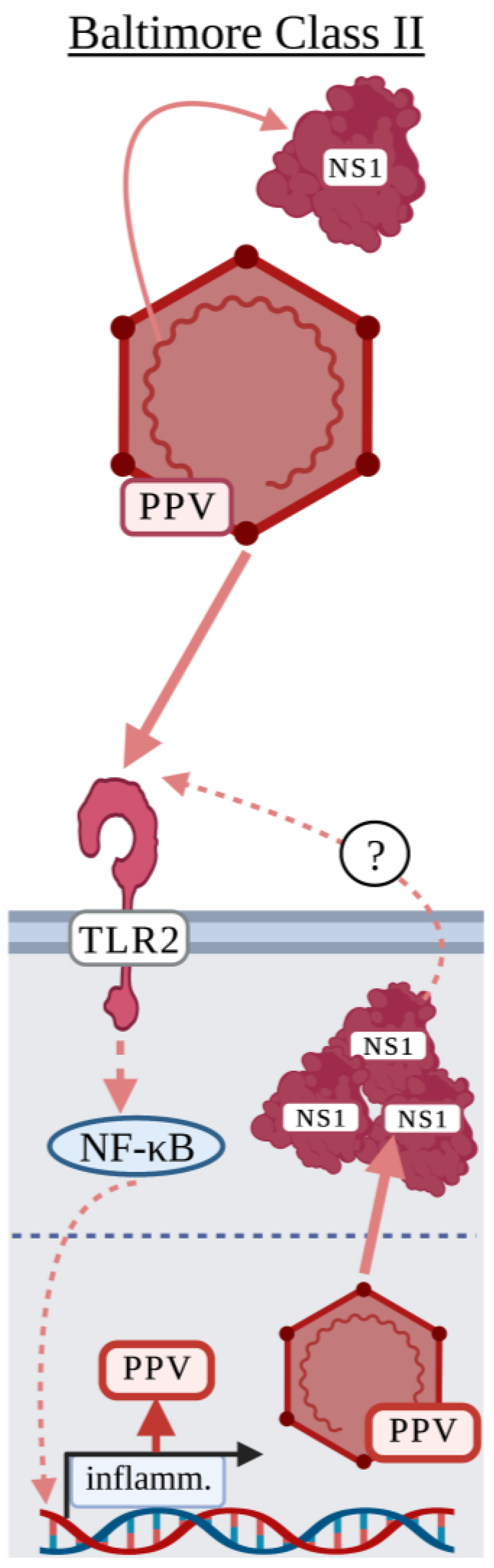
3.2. Baltimore Classification: Class II
Parvovirus
3.3. Baltimore Classification: Class III
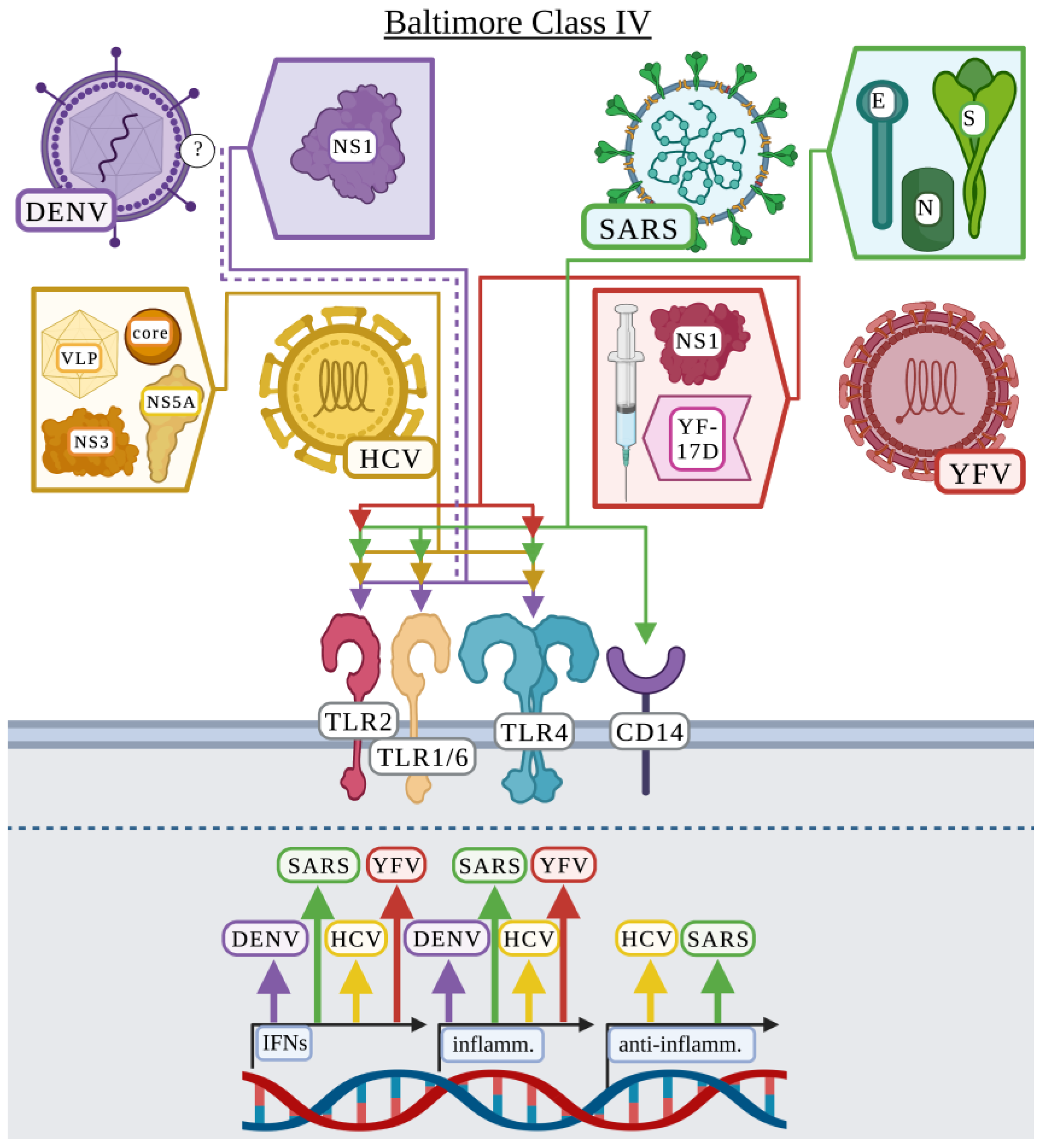
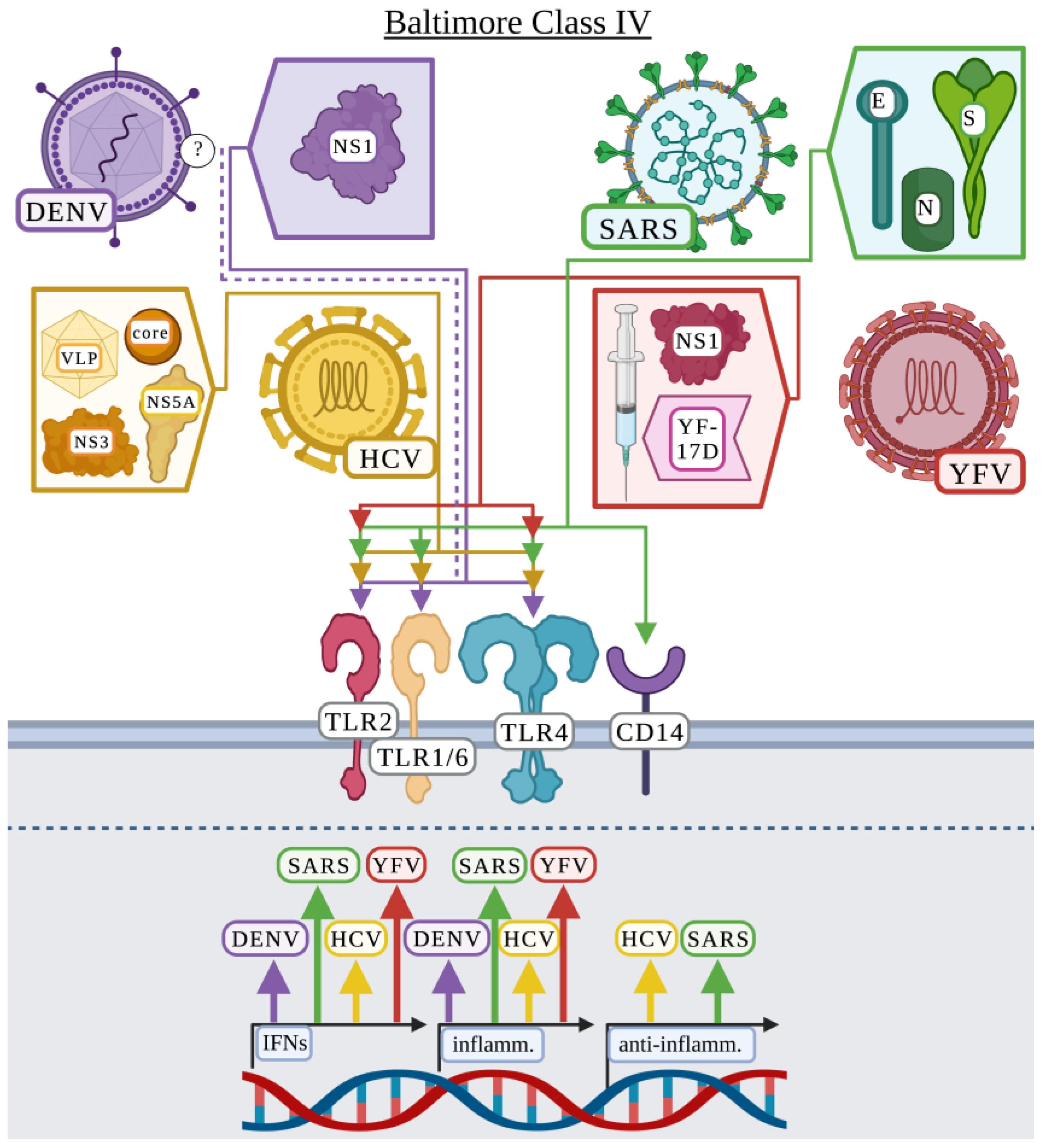
3.4. Baltimore Classification: Class IV
3.4.1. Dengue Virus
3.4.2. Hepatitis C Virus
3.4.3. Severe Acute Respiratory Syndrome Coronavirus
3.4.4. Yellow Fever Virus
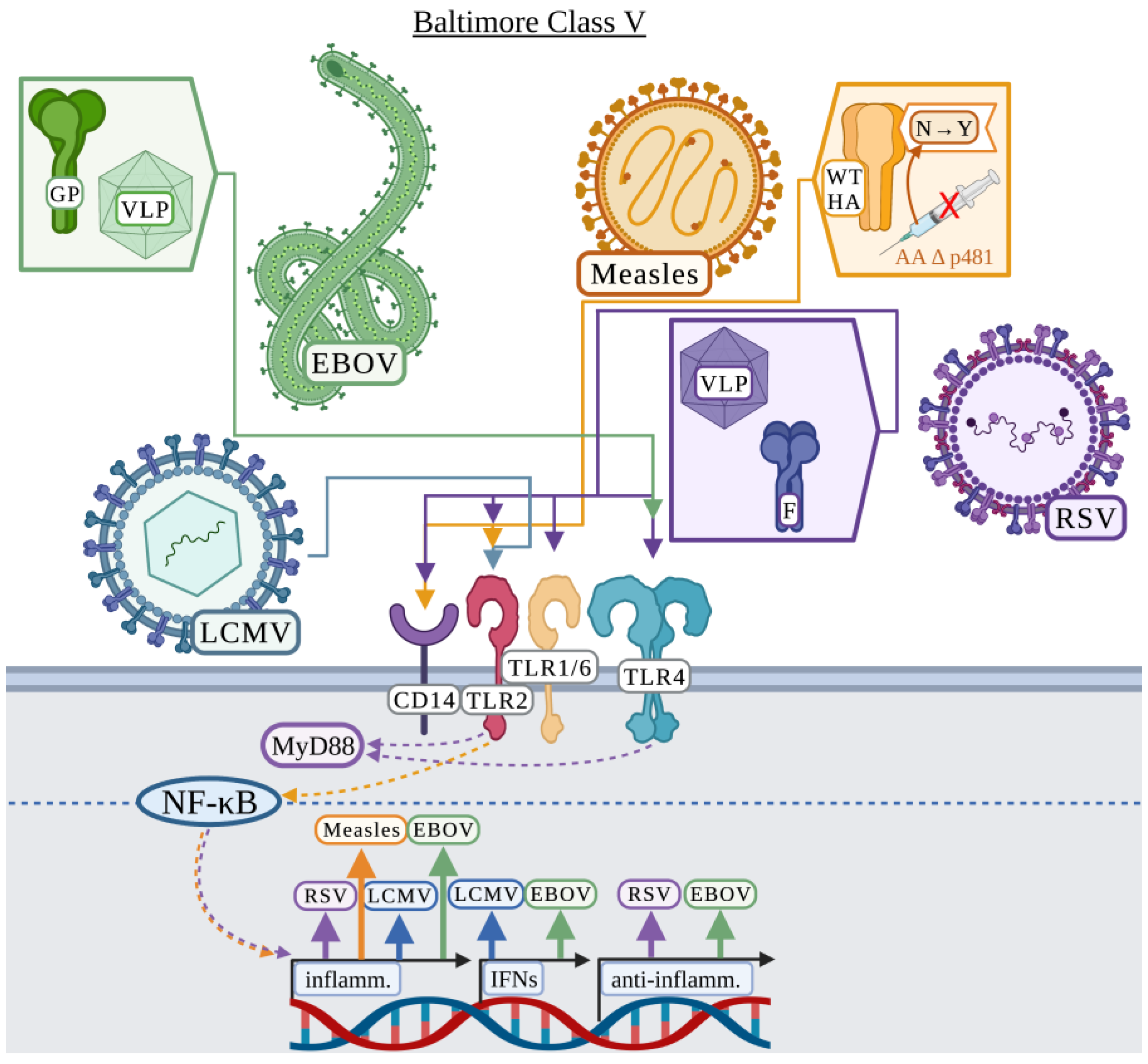
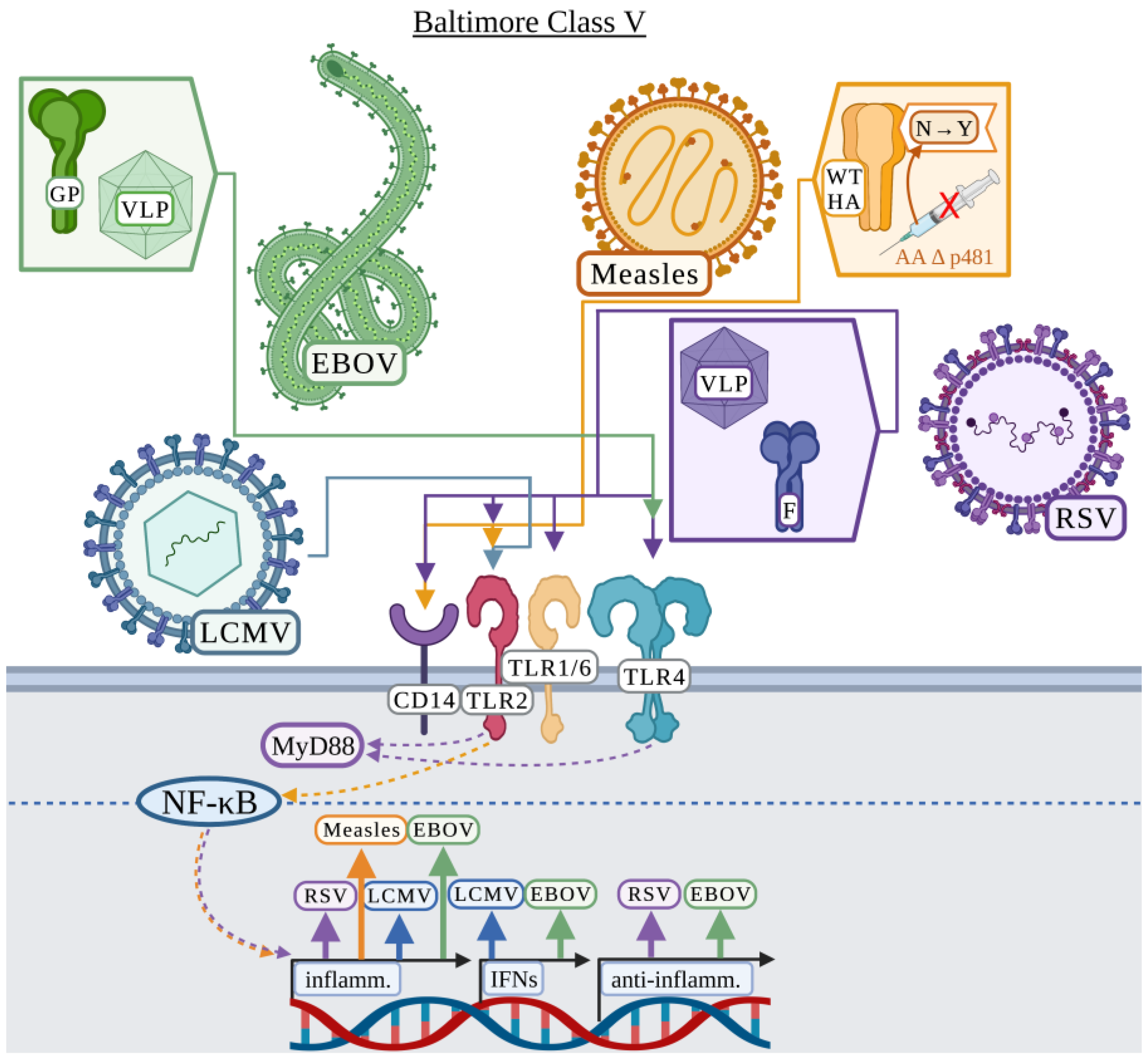
3.5. Baltimore Classification: Class V
3.5.1. Ebola Virus
3.5.2. Lymphocytic Choriomeningitis
3.5.3. Measles
3.5.4. Respiratory Syncytial Virus
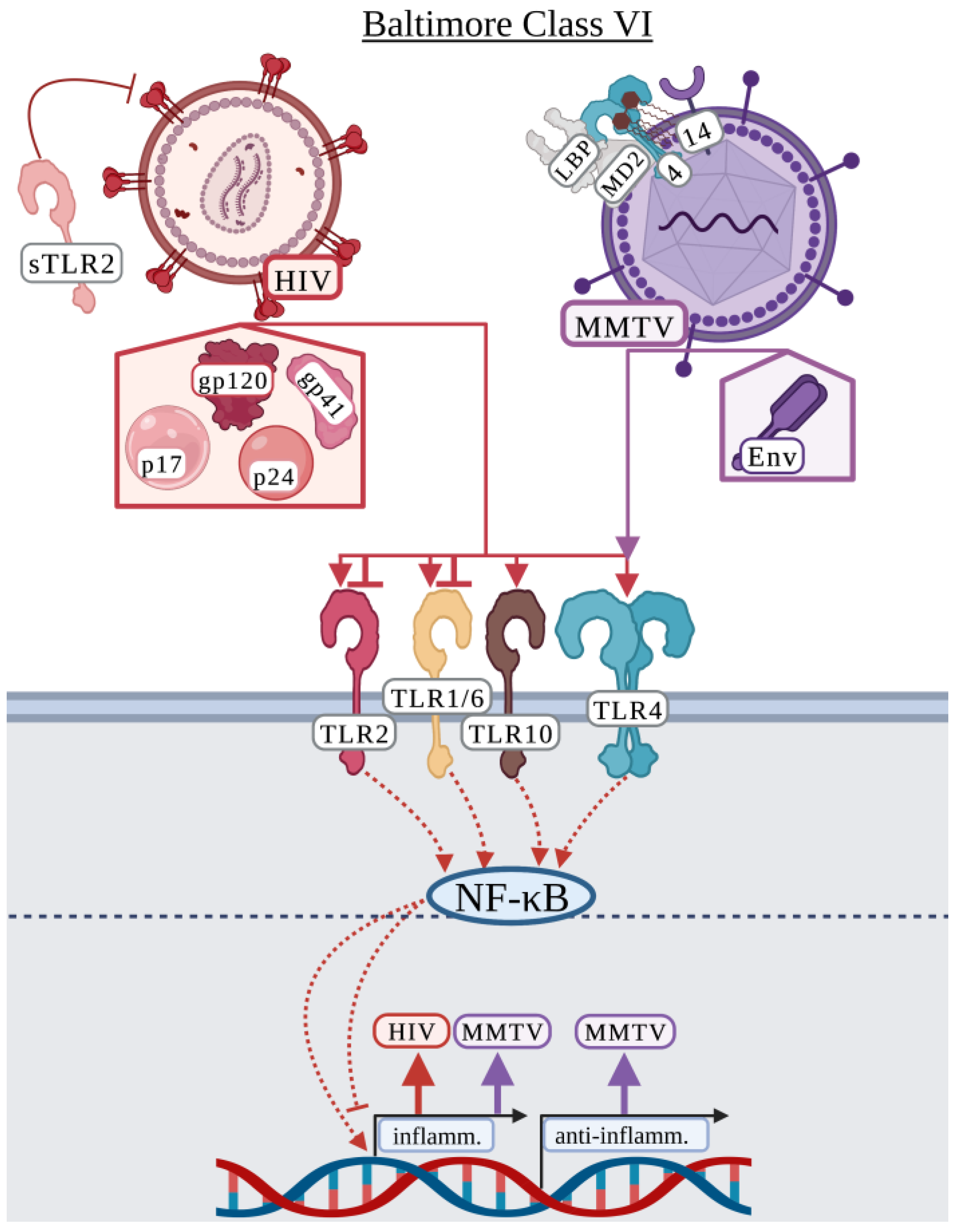
3.6. Baltimore Classification: Class VI
3.6.1. Human Immunodeficiency Virus
3.6.2. Mouse Mammary Tumor Virus
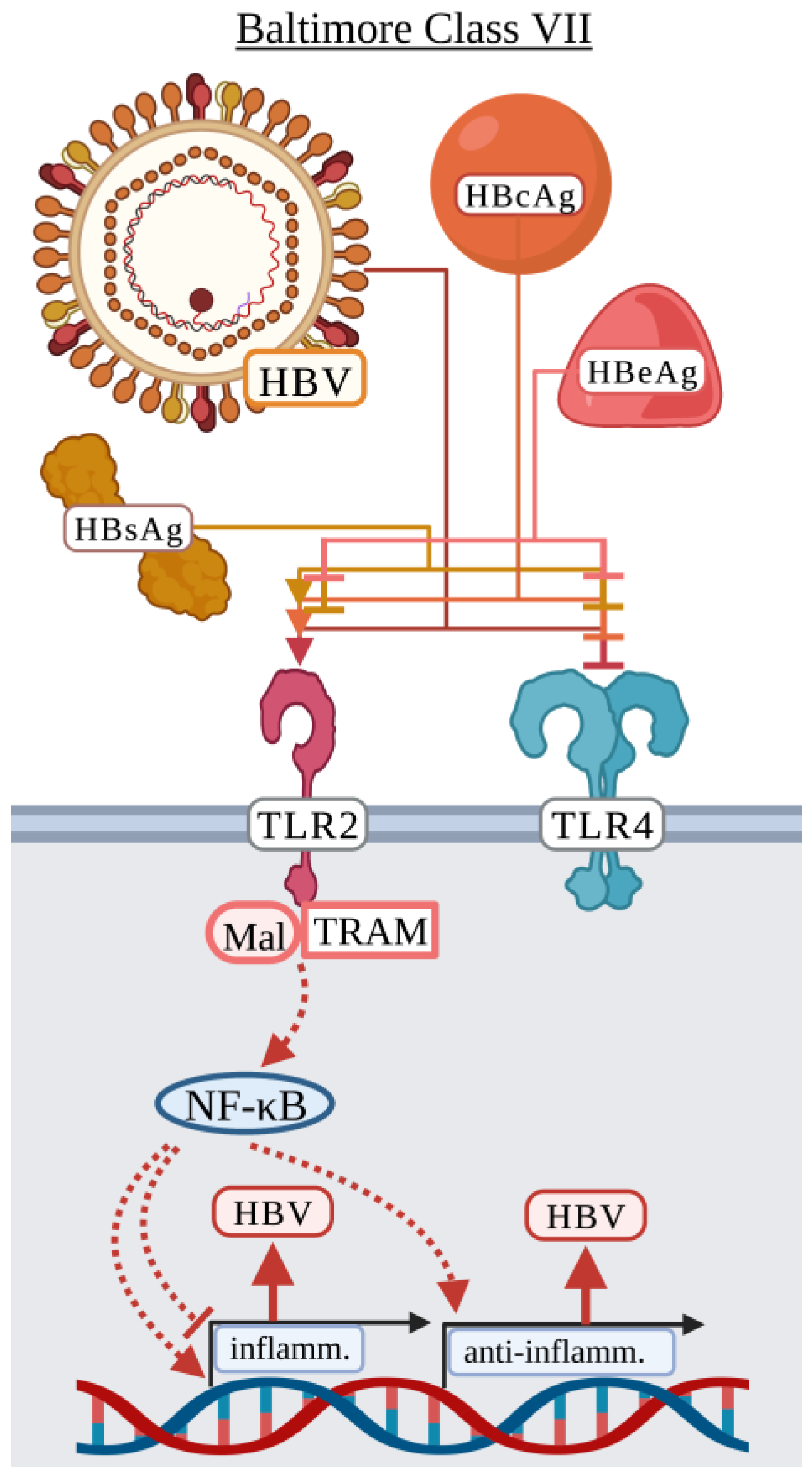
3.7. Baltimore Classification: Class VII
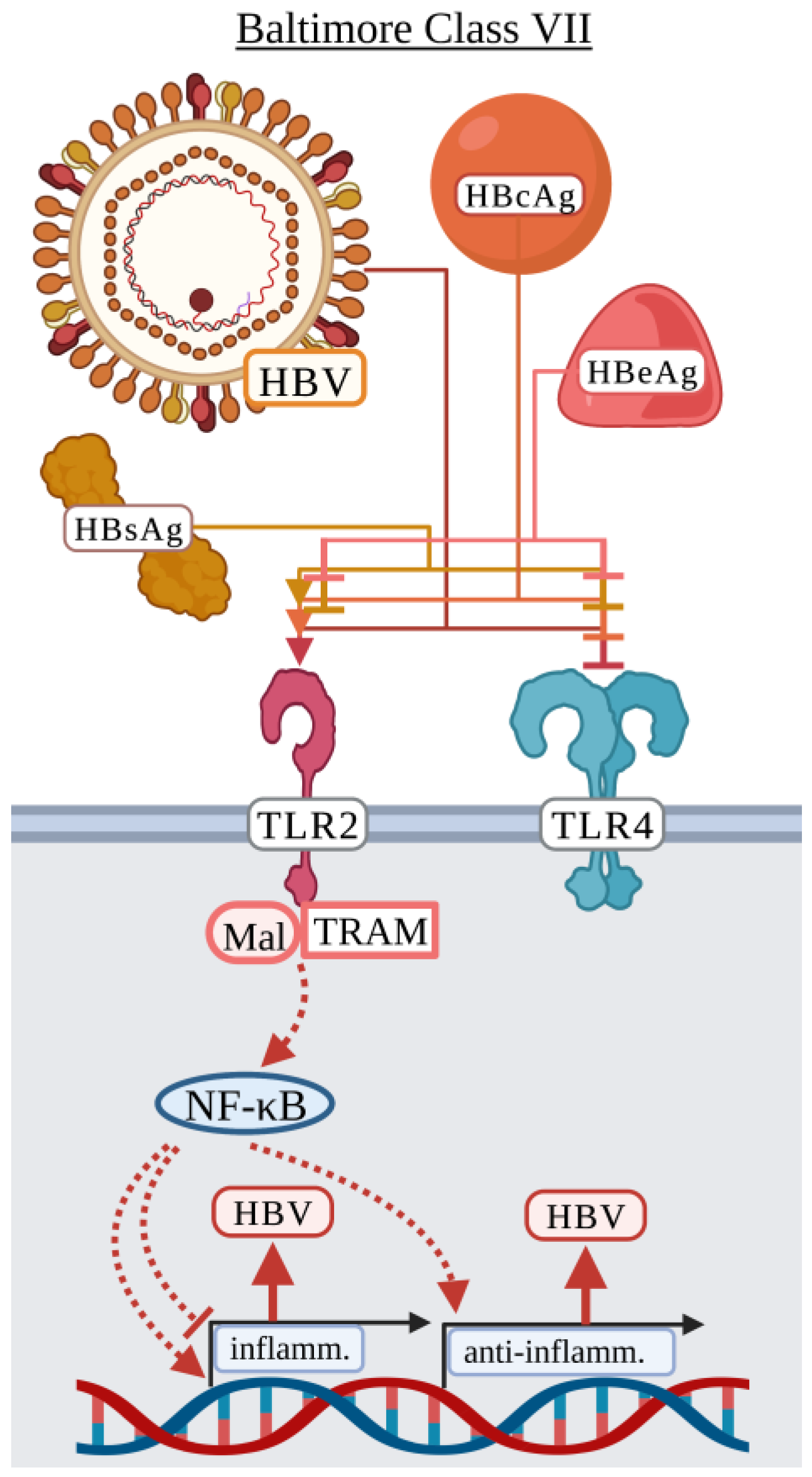
Hepatitis B Virus
4. Viruses at Toll Crossroads
5. Surface TLRs at the Forefront of Viral Pathogenesis
6. Surface TLR Promiscuity or Sending Mixed Ligand Messages
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette Spätzle/Toll/Cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Buchta, C.M.; Bishop, G.A. Toll-like Receptors and B Cells: Functions and Mechanisms. Immunol. Res. 2014, 59, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Hasan, U.; Chaffois, C.; Gaillard, C.; Saulnier, V.; Merck, E.; Tancredi, S.; Guiet, C.; Brière, F.; Vlach, J.; Lebecque, S.; et al. Human TLR10 Is a Functional Receptor, Expressed by B Cells and Plasmacytoid Dendritic Cells, which Activates Gene Transcription through MyD88. J. Immunol. 2005, 174, 2942–2950. [Google Scholar] [CrossRef] [Green Version]
- Godfroy, J.I.; Roostan, M.; Moroz, Y.S.; Korendovych, I.V.; Yin, H. Isolated Toll-like Receptor Transmembrane Domains Are Capable of Oligomerization. PLoS ONE 2012, 7, e48875. [Google Scholar] [CrossRef]
- van Bergenhenegouwen, J.; Plantinga, T.S.; Joosten, L.A.B.; Netea, M.G.; Folkerts, G.; Kraneveld, A.D.; Garssen, J.; Vos, A.P. TLR2 & Co: A Critical Analysis of the Complex Interactions between TLR2 and Coreceptors. J. Leukoc. Biol. 2013, 94, 885–902. [Google Scholar] [CrossRef]
- Ariza, M.-E.; Glaser, R.; Kaumaya, P.T.P.; Jones, C.; Williams, M.V. The EBV-Encoded DUTPase Activates NF-ΚB through the TLR2 and MyD88-Dependent Signaling Pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr Virus Encoded DUTPase Containing Exosomes Modulate Innate and Adaptive Immune Responses in Human Dendritic Cells and Peripheral Blood Mononuclear Cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human Cytomegalovirus Activates Inflammatory Cytokine Responses via CD14 and Toll-Like Receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human Cytomegalovirus Envelope Glycoproteins B and H Are Necessary for TLR2 Activation in Permissive Cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.A.; Gralewski, J.H.; Razonable, R.R. The R753Q Polymorphism Abrogates Toll-like Receptor 2 Signaling in Response to Human Cytomegalovirus. Clin. Infect. Dis. 2009, 49, e96–e99. [Google Scholar] [CrossRef] [Green Version]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.Y.; Bose, S. TLR2/MyD88/NF-ΚB Pathway, Reactive Oxygen Species, Potassium Efflux Activates NLRP3/ASC Inflammasome during Respiratory Syncytial Virus Infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin Protein of Wild-Type Measles Virus Activates Toll-Like Receptor 2 Signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [Green Version]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C Core and Nonstructural 3 Proteins Trigger Toll-like Receptor 2-Mediated Pathways and Inflammatory Activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like Receptors 1 and 6 Are Involved in TLR2-Mediated Macrophage Activation by Hepatitis C Virus Core and NS3 Proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef]
- Hoffmann, M.; Zeisel, M.B.; Jilg, N.; Paranhos-Baccalà, G.; Stoll-Keller, F.; Wakita, T.; Hafkemeyer, P.; Blum, H.E.; Barth, H.; Henneke, P.; et al. Toll-like Receptor 2 Senses Hepatitis C Virus Core Protein but Not Infectious Viral Particles. J. Innate. Immun. 2009, 1, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Watanabe, T.; Kudo, M.; Chiba, T. Hepatitis C Virus Core Protein Induces Homotolerance and Cross-Tolerance to Toll-like Receptor Ligands by Activation of Toll-like Receptor 2. J. Infect. Dis. 2010, 202, 853–861. [Google Scholar] [CrossRef]
- Swaminathan, G.; Pascual, D.; Rival, G.; Perales-Linares, R.; Martin-Garcia, J.; Navas-Martin, S. Hepatitis C Virus Core Protein Enhances HIV-1 Replication in Human Macrophages through TLR2, JNK, and MEK1/2-Dependent Upregulation of TNF-α and IL-6. FEBS Lett. 2014, 588, 3501–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajalakshmy, A.R.; Malathi, J.; Madhavan, H.N. Hepatitis C Virus NS3 Mediated Microglial Inflammation via TLR2/TLR6 MyD88/NF-ΚB Pathway and Toll like Receptor Ligand Treatment Furnished Immune Tolerance. PLoS ONE 2015, 10, e0125419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes Simplex Virus 1 Interaction with Toll-like Receptor 2 Contributes to Lethal Encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Belko, J.; Yu, C.; Newburger, P.E.; Wang, J.; Chan, M.; Knipe, D.M.; Finberg, R.W. The Role of Toll-like Receptors in Herpes Simplex Infection in Neonates. J. Infect. Dis. 2005, 191, 746–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochud, P.Y.; Magaret, A.S.; Koelle, D.M.; Aderem, A.; Wald, A. Polymorphisms in TLR2 Are Associated with Increased Viral Shedding and Lesional Rate in Patients with Genital Herpes Simplex Virus Type 2 Infection. J. Infect. Dis. 2007, 196, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Sarangi, P.P.; Kim, B.; Kurt-Jones, E.; Rouse, B.T. Innate Recognition Network Driving Herpes Simplex Virus-Induced Corneal Immunopathology: Role of the Toll Pathway in Early Inflammatory Events in Stromal Keratitis. J. Virol. 2007, 81, 11128–11138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of Specific Innate Immune Responses in Herpes Simplex Virus Infection of the Central Nervous System. J. Virol. 2012, 86, 2273–2281. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Luo, T.; Wu, F.; Mei, Y.W.; Peng, J.; Liu, H.; Li, H.R.; Zhang, S.L.; Dong, J.H.; Fang, Y.; et al. Involvement of TLR2 and TLR9 in the Anti-Inflammatory Effects of Chlorogenic Acid in HSV-1-Infected Microglia. Life Sci. 2015, 127, 12–18. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Hu, S.; Rowen, T.N.; Palmquist, J.M.; Lokensgard, J.R. Cutting Edge: TLR2-Mediated Proinflammatory Cytokine and Chemokine Production by Microglial Cells in Response to Herpes Simplex Virus. J. Immunol. 2005, 175, 4189–4193. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual Recognition of Herpes Simplex Viruses by TLR2 and TLR9 in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17343–17348. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Hu, S.; Lokensgard, J.R. Toll-like Receptor 2 Signaling Is a Mediator of Apoptosis in Herpes Simplex Virus-Infected Microglia. J. Neuroinflammation 2007, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; el Bassam, S.; Cordeiro, P.; Menezes, J. Requirement of TLR2-Mediated Signaling for the Induction of IL-15 Gene Expression in Human Monocytic Cells by HSV-1. Blood 2008, 112, 2360–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, L.N.; Reinert, L.S.; Malmgaard, L.; Bartholdy, C.; Thomsen, A.R.; Paludan, S.R. TLR2 and TLR9 Synergistically Control Herpes Simplex Virus Infection in the Brain. J. Immunol. 2008, 181, 8604–8612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes Simplex Virus Glycoproteins GH/GL and GB Bind Toll-Like Receptor 2, and Soluble GH/GL Is Sufficient To Activate NF-ΚB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Li, M.; Wang, K.; Wang, S.; Lu, Q.; Yan, J.; Mossman, K.L.; Lin, R.; Zheng, C. The Herpes Simplex Virus 1-Encoded Envelope Glycoprotein B Activates NF-ΚB through the Toll-Like Receptor 2 and MyD88/TRAF6-Dependent Signaling Pathway. PLoS ONE 2013, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.E.; Glaser, R.; Williams, M.V. Human Herpesviruses-1 Encoded DUTPases: A Family of Proteins that Modulate Dendritic Cell Function and Innate Immunity. Front. Microbiol. 2014, 5, 504. [Google Scholar] [CrossRef] [Green Version]
- Lucinda, N.; Figueiredo, M.M.; Pessoa, N.L.; Santos, B.S.Á.D.S.; Lima, G.K.; Freitas, A.M.; Machado, A.M.V.; Kroon, E.G.; Antonelli, L.R.D.V.; Campos, M.A. Dendritic Cells, Macrophages, NK and CD8+ T Lymphocytes Play Pivotal Roles in Controlling HSV-1 in the Trigeminal Ganglia by Producing IL1-Beta, INOS and Granzyme B. Virol. J. 2017, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Uyangaa, E.; Choi, J.Y.; Patil, A.M.; Hossain, F.M.A.; Park, S.O.K.; Kim, B.; Kim, K.; Eo, S.K. Dual TLR2/9 Recognition of Herpes Simplex Virus Infection Is Required for Recruitment and Activation of Monocytes and NK Cells and Restriction of Viral Dissemination to the Central Nervous System. Front. Immunol. 2018, 9, 905. [Google Scholar] [CrossRef]
- Frank, M.G.; Nguyen, K.H.; Ball, J.B.; Hopkins, S.; Kelley, T.; Baratta, M.V.; Fleshner, M.; Maier, S.F. SARS-CoV-2 Spike S1 Subunit Induces Neuroinflammatory, Microglial and Behavioral Sickness Responses: Evidence of PAMP-like Properties. Brain Behav. Immun. 2022, 100, 267–277. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 Senses the SARS-CoV-2 Envelope Protein to Produce Inflammatory Cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Planès, R.; Bert, J.-B.; Tairi, S.; BenMohamed, L.; Bahraoui, E. SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions. Viruses 2022, 14, 999. [Google Scholar] [CrossRef] [PubMed]
- van der Sluis, R.M.; Cham, L.B.; Gris-Oliver, A.; Gammelgaard, K.R.; Pedersen, J.G.; Idorn, M.; Ahmadov, U.; Hernandez, S.S.; Cémalovic, E.; Godsk, S.H.; et al. TLR2 and TLR7 Mediate Distinct Immunopathological and Antiviral Plasmacytoid Dendritic Cell Responses to SARS-CoV-2 Infection. EMBO J. 2022, 41, e109622. [Google Scholar] [CrossRef]
- Jin, X.; Yuan, Y.; Zhang, C.; Zhou, Y.; Song, Y.; Wei, Z.; Zhang, G. Porcine Parvovirus Nonstructural Protein NS1 Activates NF-ΚB and It Involves TLR2 Signaling Pathway. J. Vet. Sci. 2020, 21, e50. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Jin, X.; Zhang, C.; Liao, H.; Wang, P.; Zhou, Y.; Song, Y.; Xia, L.; Wang, L. TLR2-Mediated NF-ΚB Signaling Pathway Is Involved in PPV1-Induced Apoptosis in PK-15 Cells. Vet. Res. Commun. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ponterio, E.; Mariotti, S.; Tabolacci, C.; Ruggeri, F.M.; Nisini, R. Virus like Particles of GII.4 Norovirus Bind Toll Like Receptors 2 and 5. Immunol. Lett. 2019, 215, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Jochum, C.; Voth, R.; Rossol, S.; Meyer zum Büschenfelde, K.H.; Hess, G.; Will, H.; Schröder, H.C.; Steffen, R.; Müller, W.E. Immunosuppressive Function of Hepatitis B Antigens in Vitro: Role of Endoribonuclease V as One Potential Trans Inactivator for Cytokines in Macrophages and Human Hepatoma Cells. J. Virol. 1990, 64, 1956–1963. [Google Scholar] [CrossRef] [Green Version]
- Vanlandschoot, P.; van Houtte, F.; Roobrouck, A.; Farhoudi, A.; Leroux-Roels, G. Hepatitis B Virus Surface Antigen Suppresses the Activation of Monocytes through Interaction with a Serum Protein and a Monocyte-Specific Receptor. J. Gen. Virol. 2002, 83, 1281–1289. [Google Scholar] [CrossRef]
- Cheng, J.; Imanishi, H.; Morisaki, H.; Liu, W.; Nakamura, H.; Morisaki, T.; Hada, T. Recombinant HBsAg Inhibits LPS-Induced COX-2 Expression and IL-18 Production by Interfering with the NFκB Pathway in a Human Monocytic Cell Line, THP-1. J. Hepatol. 2005, 43, 465–471. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B Virus Surface Antigen Selectively Inhibits TLR2 Ligand–Induced IL-12 Production in Monocytes/Macrophages by Interfering with JNK Activation. J. Immunol. 2013, 190, 5142–5151. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern Recognition Receptors TLR4 and CD14 Mediate Response to Respiratory Syncytial Virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.H.; Sung, V.M.-H.; Levine, A.M.; Foung, S.; Lai, M.M.C. Hepatitis C Virus Induces Toll-Like Receptor 4 Expression, Leading to Enhanced Production of Beta Interferon and Interleukin-6. J. Virol. 2006, 80, 866–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Peng, J.; Jabbar, I.A.; Liu, X.; Filgueira, L.; Frazer, I.H.; Thomas, R. Activation of Dendritic Cells by Human Papillomavirus-like Particles through TLR4 and NF-KappaB-Mediated Signalling, Moderated by TGF-Beta. Immunol. Cell Biol. 2005, 83, 83–91. [Google Scholar] [CrossRef]
- Yang, R.; Murillo, F.M.; Delannoy, M.J.; Blosser, R.L.; Yutzy, W.H.; Uematsu, S.; Takeda, K.; Akira, S.; Viscidi, R.P.; Roden, R.B.S. B Lymphocyte Activation by Human Papillomavirus-like Particles Directly Induces Ig Class Switch Recombination via TLR4-MyD88. J. Immunol. 2005, 174, 7912–7919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola Virus Glycoprotein and Host Toll-Like Receptor 4 Leads to Induction of Proinflammatory Cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. Toll-like Receptors in Innate Immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, E.; Ding, J.L.; Byrne, B. SARM Modulates MyD88-Mediated TLR Activation through BB-Loop Dependent TIR-TIR Interactions. Biochim. Biophys. Acta 2016, 1863, 244–253. [Google Scholar] [CrossRef]
- Dietrich, N.; Lienenklaus, S.; Weiss, S.; Gekara, N.O. Murine Toll-Like Receptor 2 Activation Induces Type I Interferon Responses from Endolysosomal Compartments. PLoS ONE 2010, 5, e10250. [Google Scholar] [CrossRef] [Green Version]
- Stack, J.; Doyle, S.L.; Connolly, D.J.; Reinert, L.S.; O’Keeffe, K.M.; McLoughlin, R.M.; Paludan, S.R.; Bowie, A.G. TRAM Is Required for TLR2 Endosomal Signaling to Type I IFN Induction. J. Immunol. 2014, 193, 6090–6102. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 In Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef]
- Caplan, I.F.; Maguire-Zeiss, K.A. Toll-like Receptor 2 Signaling and Current Approaches for Therapeutic Modulation in Synucleinopathies. Front. Pharmacol. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Baltimore, D. Expression of Animal Virus Genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Agol, V.I. The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution? Microbiol. Mol. Biol. Rev. 2021, 85, e0005321. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Reeves, M. Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhao, X.Y. Human Cytomegalovirus Primary Infection and Reactivation: Insights From Virion-Carried Molecules. Front. Microbiol. 2020, 11, 1511. [Google Scholar] [CrossRef]
- Dumortier, J.; Streblow, D.N.; Moses, A.V.; Jacobs, J.M.; Kreklywich, C.N.; Camp, D.; Smith, R.D.; Orloff, S.L.; Nelson, J.A. Human Cytomegalovirus Secretome Contains Factors That Induce Angiogenesis and Wound Healing. J. Virol. 2008, 82, 6524–6535. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the Human Cytomegalovirus. Vaccine 2019, 37, 7437–7442. [Google Scholar] [CrossRef]
- Kijpittayarit, S.; Eid, A.J.; Brown, R.A.; Paya, C.V.; Razonable, R.R. Relationship between Toll-like Receptor 2 Polymorphism and Cytomegalovirus Disease after Liver Transplantation. Clin. Infect. Dis. 2007, 44, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2020, 11, 587380. [Google Scholar] [CrossRef]
- Aubrecht, T.G.; Weil, Z.M.; Ariza, M.E.; Williams, M.; Reader, B.F.; Glaser, R.; Sheridan, J.F.; Nelson, R.J. Epstein-Barr Virus (EBV)-Encoded DUTPase and Chronic Restraint Induce Impaired Learning and Memory and Sickness Responses. Physiol. Behav. 2014, 137, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr Virus Induces MCP-1 Secretion by Human Monocytes via TLR2. J. Virol. 2007, 81, 8016–8024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 Contributes to the Recognition of EBV by Primary Monocytes and Plasmacytoid Dendritic Cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Qin, Z.; Ye, Q.; Chen, P.; Wang, Z.; Yan, Q.; Luo, Z.; Liu, X.; Zhou, Y.; Xiong, W.; et al. Lactoferrin Suppresses the Epstein-Barr Virus-Induced Inflammatory Response by Interfering with Pattern Recognition of TLR2 and TLR9. Lab. Investig. 2014, 94, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell Infect. Microbiol. 2021, 10, 617578. [Google Scholar] [CrossRef]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes Simplex Virus: Global Infection Prevalence and Incidence Estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Padgett, D.A.; Sheridan, J.F.; Dorne, J.; Berntson, G.G.; Candelora, J.; Glaser, R. Social Stress and the Reactivation of Latent Herpes Simplex Virus Type 1. Proc. Natl. Acad. Sci. USA 1998, 95, 7231–7235. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Geng, S.; Suo, Y.; Wei, X.; Cai, Q.; Wu, B.; Zhou, X.; Shi, Y.; Wang, B. Critical Role of Regulatory T Cells in the Latency and Stress-Induced Reactivation of HSV-1. Cell Rep. 2018, 25, 2379–2389.e3. [Google Scholar] [CrossRef] [Green Version]
- Hilterbrand, A.T.; Heldwein, E.E. Go Go Gadget Glycoprotein!: HSV-1 Draws on Its Sizeable Glycoprotein Tool Kit to Customize Its Diverse Entry Routes. PLoS Pathog. 2019, 15, e1007660. [Google Scholar] [CrossRef]
- Brun, P.; Scarpa, M.; Marchiori, C.; Conti, J.; Kotsafti, A.; Porzionato, A.; de Caro, R.; Scarpa, M.; Calistri, A.; Castagliuolo, I. Herpes Simplex Virus Type 1 Engages Toll like Receptor 2 to Recruit Macrophages during Infection of Enteric Neurons. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mansur, D.S.; Kroon, E.G.; Nogueira, M.L.; Arantes, R.M.E.; Rodrigues, S.C.O.; Akira, S.; Gazzinelli, R.T.; Campos, M.A. Lethal Encephalitis in Myeloid Differentiation Factor 88-Deficient Mice Infected with Herpes Simplex Virus 1. Am. J. Pathol. 2005, 166, 1419–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Chen, K.; Feng, W.; Wu, X.; Li, H. TLR4-MyD88/Mal-NF-KB Axis Is Involved in Infection of HSV-2 in Human Cervical Epithelial Cells. PLoS ONE 2013, 8, e80327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Chen, K.; Feng, W.; Guo, J.; Li, H. HSV-2 Increases TLR4-Dependent Phosphorylated IRFs and IFN-β Induction in Cervical Epithelial Cells. PLoS ONE 2014, 9, e94806. [Google Scholar] [CrossRef]
- Lv, X.; Wang, H.; Su, A.; Xu, S.; Chu, Y. Herpes Simplex Virus Type 2 Infection Triggers AP-1 Transcription Activity through TLR4 Signaling in Genital Epithelial Cells. Virol. J. 2018, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Laval, K.; van Cleemput, J.; Vernejoul, J.B.; Enquist, L.W. Alphaherpesvirus Infection of Mice Primes PNS Neurons to an Inflammatory State Regulated by TLR2 and Type I IFN Signaling. PLoS Pathog. 2019, 15, e1008087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reske, A.; Pollara, G.; Krummenacher, C.; Katz, D.R.; Chain, B.M. Glycoprotein-Dependent and TLR2-Independent Innate Immune Recognition of Herpes Simplex Virus-1 by Dendritic Cells. J. Immunol. 2008, 180, 7525–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Osborne, N.R.; Zeng, W.; Donaghy, H.; McKinnon, K.; Jackson, D.C.; Cunningham, A.L. Herpes Simplex Virus Antigens Directly Activate NK Cells via TLR2, Thus Facilitating Their Presentation to CD4 T Lymphocytes. J. Immunol. 2012, 188, 4158–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 Recruits USP7 to Modulate TLR-Mediated Innate Response. Blood J. Am. Soc. Hematol. 2009, 113, 3264–3275. [Google Scholar] [CrossRef] [Green Version]
- van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes Simplex Virus Immediate-Early ICP0 Protein Inhibits Toll-Like Receptor 2-Dependent Inflammatory Responses and NF-ΚB Signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [Green Version]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes Simplex Virus US3 Tegument Protein Inhibits Toll-like Receptor 2 Signaling at or before TRAF6 Ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef]
- Lima, G.K.; Zolini, G.P.; Mansur, D.S.; Freire Lima, B.H.; Wischhoff, U.; Astigarraga, R.G.; Dias, M.F.; das Graças Almeida Silva, M.; Béla, S.R.; do Valle Antonelli, L.R.; et al. Toll-like Receptor (TLR) 2 and TLR9 Expressed in Trigeminal Ganglia Are Critical to Viral Control during Herpes Simplex Virus 1 Infection. Am. J. Pathol. 2010, 177, 2433–2445. [Google Scholar] [CrossRef] [PubMed]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.-A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2020, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human Papillomavirus Molecular Biology and Disease Association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.E.; Oxman, M.N.; et al. Varicella Zoster Virus Infection. Nat. Rev. Dis. Primers. 2015, 1, 15016. [Google Scholar] [CrossRef] [Green Version]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular Mechanisms of Varicella Zoster Virus Pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.P.; Kurt-Jones, E.A.; Shin, O.S.; Manchak, M.D.; Levin, M.J.; Finberg, R.W. Varicella-Zoster Virus Activates Inflammatory Cytokines in Human Monocytes and Macrophages via Toll-Like Receptor 2. J. Virol. 2005, 79, 12658–12666. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-R.; Huang, H.-C.; Kuo, H.-C.; Sheen, J.-M.; Ou, C.-Y.; Hsu, T.-Y.; Yang, K.D. IFN-α Production by Human Mononuclear Cells Infected with Varicella-Zoster Virus through TLR9-Dependent and -Independent Pathways. Cell Mol. Immunol. 2011, 8, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Heegaard, E.D.; Brown, K.E. Human Parvovirus B19. Clin. Microbiol. Rev. 2002, 15, 485–505. [Google Scholar] [CrossRef] [Green Version]
- Streck, A.F.; Truyen, U. Porcine Parvovirus. Curr. Issues Mol. Biol. 2020, 37, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Eledge, M.R.; Zita, M.D.; Boehme, K.W. Reovirus: Friend and Foe. Curr. Clin. Microbiol. Rep. 2019, 6, 132–138. [Google Scholar] [CrossRef]
- Ge, Y.; Mansell, A.; Ussher, J.E.; Brooks, A.E.S.; Manning, K.; Wang, C.J.H.; Taylor, J.A. Rotavirus NSP4 Triggers Secretion of Proinflammatory Cytokines from Macrophages via Toll-Like Receptor 2. J. Virol. 2013, 87, 11160–11167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Liu, L.; Chen, W.; Qin, F.; Zhou, F.; Yang, H. Ziyuglycoside II Inhibits Rotavirus Induced Diarrhea Possibly via TLR4/NF-ΚB Pathways. Biol. Pharm. Bull. 2020, 43, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Estes, M.K. Rotavirus Vaccines and Pathogenesis: 2008. Curr. Opin. Gastroenterol. 2009, 25, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue Infection. Nat. Rev. Dis. Primers. 2016, 2, 1–25. [Google Scholar] [CrossRef]
- Park, J.; Kim, J.; Jang, Y.S. Current Status and Perspectives on Vaccine Development against Dengue Virus Infection. J. Microbiol. 2022, 60, 247–254. [Google Scholar] [CrossRef]
- Chen, J.; Ng, M.M.-L.; Chu, J.J.H. Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection. PLoS Pathog. 2015, 11, e1005053. [Google Scholar] [CrossRef]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue Virus NS1 Protein Activates Cells via Toll-like Receptor 4 and Disrupts Endothelial Cell Monolayer Integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [Green Version]
- Modhiran, N.; Watterson, D.; Blumenthal, A.; Baxter, A.G.; Young, P.R.; Stacey, K.J. Dengue Virus NS1 Protein Activates Immune Cells via TLR4 but Not TLR2 or TLR6. Immunol. Cell Biol. 2017, 95, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ng, M.M.L.; Chu, J.J.H. Molecular Profiling of T-Helper Immune Genes during Dengue Virus Infection. Virol. J. 2008, 5, 165. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Briseño, J.A.; Upasani, V.; Ellen, B.M.T.; Moser, J.; Pauzuolis, M.; Ruiz-Silva, M.; Heng, S.; Laurent, D.; Choeung, R.; Dussart, P.; et al. TLR2 on Blood Monocytes Senses Dengue Virus Infection and Its Expression Correlates with Disease Pathogenesis. Nat. Commun. 2020, 11, 3177. [Google Scholar] [CrossRef]
- Aguilar Briseño, J.A.; Ramos Pereira, L.; van der Laan, M.; Pauzuolis, M.; ter Ellen, B.M.; Upasani, V.; Moser, J.; de Souza Ferreira, L.C.; Smit, J.M.; Rodenhuis-Zybert, I.A. TLR2 Axis on Peripheral Blood Mononuclear Cells Regulates Inflammatory Responses to Non-Infectious Immature Dengue Virus Particles. PLoS Pathog. 2022, 18, e1010499. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.-M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C Virus Infection. Nat. Rev. Dis. Primers. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Morozov, V.A.; Lagaye, S. Hepatitis C Virus: Morphogenesis, Infection and Therapy. World J. Hepatol. 2018, 10, 186–212. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, E.; Kiyosawa, K.; Matsumoto, A.; Kashiwakuma, T.; Hasegawa, A.; Mori, H.; Yanagihara, O.; Ohta, Y. Serum Levels of Hepatitis C Virus Core Protein in Patients With Chronic Hepatitis C Treated with Interferon Alfa. Hepatology 1996, 23, 1330–1333. [Google Scholar] [CrossRef]
- Karamichali, E.; Chihab, H.; Kakkanas, A.; Marchio, A.; Karamitros, T.; Pogka, V.; Varaklioti, A.; Kalliaropoulos, A.; Martinez-Gonzales, B.; Foka, P.; et al. HCV Defective Genomes Promote Persistent Infection by Modulating the Viral Life Cycle. Front. Microbiol. 2018, 9, 2942. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Zhai, N.; Song, H.; Li, H.; Yang, Y.; Li, T.; Guo, X.; Chi, B.; Niu, J.; et al. HCV Core Protein Inhibits Polarization and Activity of Both M1 and M2 Macrophages through the TLR2 Signaling Pathway. Sci. Rep. 2016, 6, 36160. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- World Health Organization WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 19 September 2022).
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS Coronavirus Spike Protein-Induced Innate Immune Response Occurs via Activation of the NF-KappaB Pathway in Human Monocyte Macrophages in Vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-Mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Olajide, O.A.; Iwuanyanwu, V.U.; Adegbola, O.D.; Al-Hindawi, A.A. SARS-CoV-2 Spike Glycoprotein S1 Induces Neuroinflammation in BV-2 Microglia. Mol. Neurobiol. 2022, 59, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kizaki, T. SARS-CoV-2 Spike Protein S1 Subunit Induces pro-Inflammatory Responses via Toll-like Receptor 4 Signaling in Murine and Human Macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 Spike Protein Interacts with and Activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 Spike Protein Induces Inflammation via TLR2-Dependent Activation of the NF-ΚB Pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Furtado, N.D.; Raphael, L.d.M.; Ribeiro, I.P.; de Mello, I.S.; Fernandes, D.R.; Gómez, M.M.; dos Santos, A.A.C.; Nogueira, M.d.S.; de Castro, M.G.; de Abreu, F.V.S.; et al. Biological Characterization of Yellow Fever Viruses Isolated From Non-Human Primates in Brazil With Distinct Genomic Landscapes. Front. Microbiol. 2022, 13, 757084. [Google Scholar] [CrossRef]
- Querec, T.; Bennouna, S.; Alkan, S.; Laouar, Y.; Gorden, K.; Flavell, R.; Akira, S.; Ahmed, R.; Pulendran, B. Yellow Fever Vaccine YF-17D Activates Multiple Dendritic Cell Subsets via TLR2, 7, 8, and 9 to Stimulate Polyvalent Immunity. J. Exp. Med. 2006, 203, 413–424. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization: Ebola Situation Report-30 March 2016. Available online: https://apps.who.int/iris/bitstream/handle/10665/204714/ebolasitrep_30mar2016_eng.pdf;jsessionid=443054184BE33668ED91C4ECDA106F92?sequence=1 (accessed on 11 November 2022).
- Jacob, S.T.; Crozier, I.; Fischer, W.A.; Hewlett, A.; Kraft, C.S.; de la Vega, M.-A.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola Virus Disease. Nat. Rev. Dis. Primers. 2020, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Bharat, T.A.M.; Noda, T.; Riches, J.D.; Kraehling, V.; Kolesnikova, L.; Becker, S.; Kawaoka, Y.; Briggs, J.A.G. Structural Dissection of Ebola Virus and Its Assembly Determinants Using Cryo-Electron Tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4275–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.; Peters, C.J.; Nichol, S.T. The Virion Glycoproteins of Ebola Viruses Are Encoded in Two Reading Frames and Are Expressed through Transcriptional Editing. Proc. Natl. Acad. Sci. USA 1996, 93, 3602–3607. [Google Scholar] [CrossRef] [Green Version]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent Modifications of the Ebola Virus Glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar] [CrossRef]
- Escudero-Pérez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.; Volchkov, V.E. Shed GP of Ebola Virus Triggers Immune Activation and Increased Vascular Permeability. PLoS Pathog. 2014, 10, e1004509. [Google Scholar] [CrossRef] [PubMed]
- Iampietro, M.; Younan, P.; Nishida, A.; Dutta, M.; Lubaki, N.M.; Santos, R.I.; Koup, R.A.; Katze, M.G.; Bukreyev, A. Ebola Virus Glycoprotein Directly Triggers T Lymphocyte Death despite of the Lack of Infection. PLoS Pathog. 2017, 13, e1006397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola Virus Glycoprotein Induces an Innate Immune Response In Vivo via TLR4. Front. Microbiol. 2017, 8, 1571. [Google Scholar] [CrossRef] [Green Version]
- Younan, P.; Ramanathan, P.; Graber, J.; Gusovsky, F.; Bukreyev, A. The Toll-Like Receptor 4 Antagonist Eritoran Protects Mice from Lethal Filovirus Challenge. MBio 2017, 8, e00226-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iampietro, M.; Santos, R.I.; Lubaki, N.M.; Bukreyev, A. Ebola Virus Shed Glycoprotein Triggers Differentiation, Infection, and Death of Monocytes Through Toll-Like Receptor 4 Activation. J. Infect. Dis. 2018, 218, S327–S334. [Google Scholar] [CrossRef]
- Vilibic-Cavlek, T.; Savic, V.; Ferenc, T.; Mrzljak, A.; Barbic, L.; Bogdanic, M.; Stevanovic, V.; Tabain, I.; Ferencak, I.; Zidovec-Lepej, S. Lymphocytic Choriomeningitis—Emerging Trends of a Neglected Virus: A Narrative Review. Trop. Med. Infect. Dis. 2021, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Lapošová, K.; Pastoreková, S.; Tomášková, J. Lymphocytic Choriomeningitis Virus: Invisible but Not Innocent. Acta Virol. 2013, 57, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 Is Critical for the Development of Innate and Adaptive Immunity during Acute Lymphocytic Choriomeningitis Virus Infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic Choriomeningitis Virus (LCMV) Infection of CNS Glial Cells Results in TLR2-MyD88/Mal-Dependent Inflammatory Responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, T.; Maenaka, K.; Yanagi, Y. Measles Virus Hemagglutinin: Structural Insights into Cell Entry and Measles Vaccine. Front. Microbiol. 2011, 2, 247. [Google Scholar] [CrossRef]
- Griffin, D.E. Measles Vaccine. Viral. Immunol. 2018, 31, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Saltel, F.; Escola, J.-M.; Jurdic, P.; Wild, T.F.; Horvat, B. Cell Surface Delivery of the Measles Virus Nucleoprotein: A Viral Strategy To Induce Immunosuppression. J. Virol. 2004, 78, 11952–11961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, J.; Britton, P.N.; King, C.L.; Booy, R. The Immunogenicity and Safety of Respiratory Syncytial Virus Vaccines in Development: A Systematic Review. Influenza Other Respir. Viruses 2021, 15, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Bukreyev, A.; Yang, L.; Collins, P.L. The Secreted G Protein of Human Respiratory Syncytial Virus Antagonizes Antibody-Mediated Restriction of Replication Involving Macrophages and Complement. J. Virol. 2012, 86, 10880–10884. [Google Scholar] [CrossRef] [Green Version]
- Kiss, G.; Holl, J.M.; Williams, G.M.; Alonas, E.; Vanover, D.; Lifland, A.W.; Gudheti, M.; Guerrero-Ferreira, R.C.; Nair, V.; Yi, H.; et al. Structural Analysis of Respiratory Syncytial Virus Reveals the Position of M2-1 between the Matrix Protein and the Ribonucleoprotein Complex. J. Virol. 2014, 88, 7602–7617. [Google Scholar] [CrossRef] [Green Version]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of Toll-Like Receptor 4 in Innate Immunity to Respiratory Syncytial Virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef] [Green Version]
- Haeberle, H.A.; Takizawa, R.; Casola, A.; Brasier, A.R.; Dieterich, H.-J.; van Rooijen, N.; Gatalica, Z.; Garofalo, R.P. Respiratory Syncytial Virus-Induced Activation of Nuclear Factor-KB in the Lung Involves Alveolar Macrophages and Toll-Like Receptor 4-Dependent Pathways. J. Infect. Dis. 2002, 186, 1199–1206. [Google Scholar] [CrossRef]
- Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Butler, N.S.; Carter, A.B.; Gudmundsson, G.; Hunninghake, G.W. Respiratory Syncytial Virus Up-Regulates TLR4 and Sensitizes Airway Epithelial Cells to Endotoxin. J. Biol. Chem. 2003, 278, 53035–53044. [Google Scholar] [CrossRef] [Green Version]
- Tulic, M.K.; Hurrelbrink, R.J.; Prêle, C.M.; Laing, I.A.; Upham, J.W.; le Souef, P.; Sly, P.D.; Holt, P.G. TLR4 Polymorphisms Mediate Impaired Responses to Respiratory Syncytial Virus and Lipopolysaccharide. J. Immunol. 2007, 179, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.C.; Porto, B.N. Respiratory Syncytial Virus Fusion Protein Promotes TLR-4-Dependent Neutrophil Extracellular Trap Formation by Human Neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef] [Green Version]
- Walpita, P.; Johns, L.M.; Tandon, R.; Moore, M.L. Mammalian Cell-Derived Respiratory Syncytial Virus-like Particles Protect the Lower as Well as the Upper Respiratory Tract. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory Syncytial Virus Activates Innate Immunity through Toll-Like Receptor 2. J. Virol. 2009, 83, 1492–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marr, N.; Turvey, S.E. Role of Human TLR4 in Respiratory Syncytial Virus-Induced NF-ΚB Activation, Viral Entry and Replication. Innate. Immun. 2012, 18, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Douville, R.N.; Lissitsyn, Y.; Hirschfeld, A.F.; Becker, A.B.; Kozyrskyj, A.L.; Liem, J.; Bastien, N.; Li, Y.; Victor, R.E.; Sekhon, M.; et al. TLR4 Asp299Gly and Thr399Ile Polymorphisms: No Impact on Human Immune Responsiveness to LPS or Respiratory Syncytial Virus. PLoS ONE 2010, 5, e12087. [Google Scholar] [CrossRef] [Green Version]
- Ehl, S.; Bischoff, R.; Ostler, T.; Vallbracht, S.; Schulte-Mönting, J.; Poltorak, A.; Freudenberg, M. The Role of Toll-like Receptor 4 versus Interleukin-12 in Immunity to Respiratory Syncytial Virus. Eur. J. Immunol. 2004, 34, 1146–1153. [Google Scholar] [CrossRef]
- Triantafilou, K.; Kar, S.; Vakakis, E.; Kotecha, S.; Triantafilou, M. Human Respiratory Syncytial Virus Viroporin SH: A Viral Recognition Pathway Used by the Host to Signal Inflammasome Activation. Thorax 2013, 68, 66–75. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization Key Facts HIV. Available online: https://cdn.who.int/media/docs/default-source/hq-hiv-hepatitis-and-stis-library/key-facts-hiv-2021-26july2022.pdf?sfvrsn=8f4e7c93_5 (accessed on 18 September 2022).
- Engelman, A.; Cherepanov, P. The Structural Biology of HIV-1: Mechanistic and Therapeutic Insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Marincowitz, C.; Genis, A.; Goswami, N.; de Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular Endothelial Dysfunction in the Wake of HIV and ART. FEBS J. 2019, 286, 1256–1270. [Google Scholar] [CrossRef] [Green Version]
- German Advisory Committee Blood (Arbeitskreis Blut), S. Assessment of P.T. by B. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemother. 2016, 43, 203–222. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Vasan, S.; Kim, J.H.; Ake, J.A. Current Approaches to HIV Vaccine Development: A Narrative Review. J. Int. AIDS Soc. 2021, 24 (Suppl. S7), e25793. [Google Scholar] [CrossRef]
- Lester, R.T.; Yao, X.-D.; Ball, T.B.; McKinnon, L.R.; Kaul, R.; Wachihi, C.; Jaoko, W.; Plummer, F.A.; Rosenthal, K.L. Toll-like Receptor Expression and Responsiveness Are Increased in Viraemic HIV-1 Infection. AIDS 2008, 22, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Berzsenyi, M.D.; Roberts, S.K.; Preiss, S.; Woollard, D.J.; Beard, M.R.; Skinner, N.A.; Bowden, D.S.; Visvanathan, K. Hepatic TLR2 & TLR4 Expression Correlates with Hepatic Inflammation and TNF-α in HCV & HCV/HIV Infection. J. Viral. Hepat. 2011, 18, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.C.; Arteaga, J.; Paul, S.; Kumar, A.; Latz, E.; Urcuqui-Inchima, S. Up-Regulation of TLR2 and TLR4 in Dendritic Cells in Response to HIV Type 1 and Coinfection with Opportunistic Pathogens. AIDS Res. Hum. Retroviruses 2011, 27, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, J.C.; Stevenson, M.; Latz, E.; Urcuqui-Inchima, S. HIV Type 1 Infection Up-Regulates TLR2 and TLR4 Expression and Function in Vivo and in Vitro. AIDS Res. Hum. Retroviruses 2012, 28, 1313–1328. [Google Scholar] [CrossRef] [Green Version]
- Vidyant, S.; Chatterjee, A.; Agarwal, V.; Dhole, T.N. Susceptibility to HIV-1 Infection Is Influenced by Toll like Receptor-2 (−196 to −174) Polymorphism in a North Indian Population. J. Gene Med. 2017, 19, e2971. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Drannik, A.G.; Abimiku, A.; Rosenthal, K.L.; INFANT Study Team. Soluble Toll-like Receptor 2 Is Significantly Elevated in HIV-1 Infected Breast Milk and Inhibits HIV-1 Induced Cellular Activation, Inflammation and Infection. AIDS 2014, 28, 2023–2032. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Rosenthal, K.L.; INFANT Study Team. HIV-1 Structural Proteins Serve as PAMPs for TLR2 Heterodimers Significantly Increasing Infection and Innate Immune Activation. Front. Immunol. 2015, 6, 426. [Google Scholar] [CrossRef] [Green Version]
- Henrick, B.M.; Yao, X.D.; Zahoor, M.A.; Abimiku, A.; Osawe, S.; Rosenthal, K.L. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front. Immunol. 2019, 10, 482. [Google Scholar] [CrossRef]
- Henrick, B.M.; Nag, K.; Yao, X.D.; Drannik, A.G.; Aldrovandi, G.M.; Rosenthal, K.L. Milk Matters: Soluble Toll-like Receptor 2 (STLR2) in Breast Milk Significantly Inhibits HIV-1 Infection and Inflammation. PLoS ONE 2012, 7, e40138. [Google Scholar] [CrossRef]
- Reuven, E.M.; Ali, M.; Rotem, E.; Schwarzer, R.; Schwarzter, R.; Gramatica, A.; Futerman, A.H.; Shai, Y. The HIV-1 Envelope Transmembrane Domain Binds TLR2 through a Distinct Dimerization Motif and Inhibits TLR2-Mediated Responses. PLoS Pathog. 2014, 10, e1004248. [Google Scholar] [CrossRef]
- Nazli, A.; Kafka, J.K.; Ferreira, V.H.; Anipindi, V.; Mueller, K.; Osborne, B.J.; Dizzell, S.; Chauvin, S.; Mian, M.F.; Ouellet, M.; et al. HIV-1 Gp120 Induces TLR2- and TLR4-Mediated Innate Immune Activation in Human Female Genital Epithelium. J. Immunol. 2013, 191, 4246–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.R. Mouse Mammary Tumor Virus Molecular Biology and Oncogenesis. Viruses 2010, 2, 2000–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, M.; Case, L.K.; Kopaskie, K.; Kozlova, A.; MacDearmid, C.; Chervonsky, A.V.; Golovkina, T.V. Successful Transmission of a Retrovirus Depends on the Commensal Microbiota. Science 2011, 334, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilks, J.; Lien, E.; Jacobson, A.N.; Fischbach, M.A.; Qureshi, N.; Chervonsky, A.V.; Golovkina, T.V. Mammalian Lipopolysaccharide Receptors Incorporated into the Retroviral Envelope Augment Virus Transmission. Cell Host Microbe 2015, 18, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Rassa, J.C.; Meyers, J.L.; Zhang, Y.; Kudaravalli, R.; Ross, S.R. Murine Retroviruses Activate B Cells via Interaction with Toll-like Receptor 4. Proc. Natl. Acad. Sci. USA 2002, 99, 2281–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jude, B.A.; Pobezinskaya, Y.; Bishop, J.; Parke, S.; Medzhitov, R.M.; Chervonsky, A.V.; Golovkina, T.V. Subversion of the Innate Immune System by a Retrovirus. Nat. Immunol. 2003, 4, 573–578. [Google Scholar] [CrossRef]
- Burzyn, D.; Rassa, J.C.; Kim, D.; Nepomnaschy, I.; Ross, S.R.; Piazzon, I. Toll-like Receptor 4-Dependent Activation of Dendritic Cells by a Retrovirus. J. Virol. 2004, 78, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Courreges, M.C.; Burzyn, D.; Nepomnaschy, I.; Piazzon, I.; Ross, S.R. Critical Role of Dendritic Cells in Mouse Mammary Tumor Virus in Vivo Infection. J. Virol. 2007, 81, 3769–3777. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.-F.; Chen, D.-S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.-L. Hepatitis B Virus Infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef]
- Kramvis, A.; Kostaki, E.-G.; Hatzakis, A.; Paraskevis, D. Immunomodulatory Function of HBeAg Related to Short-Sighted Evolution, Transmissibility, and Clinical Manifestation of Hepatitis B Virus. Front. Microbiol. 2018, 9, 2521. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.-A.; Do, S.Y.; Kim, B.-J. Precore/Core Region Mutations of Hepatitis B Virus Related to Clinical Severity. World J. Gastroenterol. 2016, 22, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Bonino, F.; Colombatto, P.; Brunetto, M.R. HBeAg-Negative/Anti-HBe-Positive Chronic Hepatitis B: A 40-Year-Old History. Viruses 2022, 14, 1691. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Chen, Y.-S.; Cao, L.; Chen, X.-W.; Lu, M.-J. Hepatitis B Virus Infection: Defective Surface Antigen Expression and Pathogenesis. World J. Gastroenterol. 2018, 24, 3488–3499. [Google Scholar] [CrossRef]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.V.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of Toll-like Receptor-2 Expression in Chronic Hepatitis B by the Precore Protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The Hepatitis B e Antigen (HBeAg) Targets and Suppresses Activation of the Toll-like Receptor Signaling Pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef]
- Cooper, A.; Tal, G.; Lider, O.; Shaul, Y. Cytokine Induction by the Hepatitis B Virus Capsid in Macrophages Is Facilitated by Membrane Heparan Sulfate and Involves TLR2. J. Immunol. 2005, 175, 3165–3176. [Google Scholar] [CrossRef]
- Li, M.; Sun, R.; Xu, L.; Yin, W.; Chen, Y.; Zheng, X.; Lian, Z.; Wei, H.; Tian, Z. Kupffer Cells Support Hepatitis B Virus–Mediated CD8 + T Cell Exhaustion via Hepatitis B Core Antigen–TLR2 Interactions in Mice. J. Immunol. Immunol. 2015, 195, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.; Zhang, Y.; Yang, X.; Li, M.; Hu, H.; Xiong, J.; Wang, N.; Jin, J.; Zhang, Y.; Song, Y.; et al. Hepatitis B Core Antigen Impairs the Polarization While Promoting the Production of Inflammatory Cytokines of M2 Macrophages via the TLR2 Pathway. Front. Immunol. 2020, 11, 535. [Google Scholar] [CrossRef]
- Song, H.; Tan, G.; Yang, Y.; Cui, A.; Li, H.; Li, T.; Wu, Z.; Yang, M.; Lv, G.; Chi, X.; et al. Hepatitis B Virus–Induced Imbalance of Inflammatory and Antiviral Signaling by Differential Phosphorylation of STAT1 in Human Monocytes. J. Immunol. 2019, 202, 2266–2275. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Trippler, M.; Real, C.I.; Werner, M.; Luo, X.; Schefczyk, S.; Kemper, T.; Anastasiou, O.E.; Ladiges, Y.; Treckmann, J.; et al. Hepatitis B Virus Particles Activate Toll-Like Receptor 2 Signaling Initially Upon Infection of Primary Human Hepatocytes. Hepatology 2020, 72, 829–844. [Google Scholar] [CrossRef]
- Li, Q.; Wang, J.; Islam, H.; Kirschning, C.; Lu, H.; Hoffmann, D.; Dittmer, U.; Lu, M. Hepatitis B Virus Particles Activate B Cells through the TLR2–MyD88–MTOR Axis. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kirschning, C.J.; Wesche, H.; Merrill Ayres, T.; Rothe, M. Human Toll-like Receptor 2 Confers Responsiveness to Bacterial Lipopolysaccharide. J. Exp. Med. 1998, 188, 2091–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.B.; Mark, M.R.; Gray, A.; Huang, A.; Xie, M.H.; Zhang, M.; Goddard, A.; Wood, W.I.; Gurney, A.L.; Godowski, P.J. Toll-like Receptor-2 Mediates Lipopolysaccharide-Induced Cellular Signalling. Nature 1998, 395, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Lee, J.; Tobias, P.S. Two Lipoproteins Extracted from Escherichia Coli K-12 LCD25 Lipopolysaccharide Are the Major Components Responsible for Toll-like Receptor 2-Mediated Signaling. J. Immunol. 2002, 168, 4012–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, J.; Tehan, M.M.; Warren, H.S. Murein Lipoprotein, Peptidoglycan-Associated Lipoprotein, and Outer Membrane Protein A Are Present in Purified Rough and Smooth Lipopolysaccharides. J. Infect. Dis. 2003, 188, 286–289. [Google Scholar] [CrossRef]
- Good, D.W.; George, T.; Watts, B.A. Toll-like Receptor 2 Is Required for LPS-Induced Toll-like Receptor 4 Signaling and Inhibition of Ion Transport in Renal Thick Ascending Limb. J. Biol. Chem. 2012, 287, 20208–20220. [Google Scholar] [CrossRef] [Green Version]
- Darfour-Oduro, K.A.; Megens, H.-J.; Roca, A.; Groenen, M.A.M.; Schook, L.B. Evidence for Adaptation of Porcine Toll-like Receptors. Immunogenetics 2016, 68, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Grabiec, A.; Meng, G.; Fichte, S.; Bessler, W.; Wagner, H.; Kirschning, C.J. Human but Not Murine Toll-like Receptor 2 Discriminates between Tri-Palmitoylated and Tri-Lauroylated Peptides. J. Biol. Chem. 2004, 279, 48004–48012. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal Structure of the TLR1-TLR2 Heterodimer Induced by Binding of a Tri-Acylated Lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Düesberg, U.; von dem Bussche, A.; Kirschning, C.; Miyake, K.; Sauerbruch, T.; Spengler, U. Cell Activation by Synthetic Lipopeptides of the Hepatitis C Virus (HCV)--Core Protein Is Mediated by Toll like Receptors (TLRs) 2 and 4. Immunol. Lett. 2002, 84, 89–95. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Zähringer, U.; Lindner, B.; Inamura, S.; Heine, H.; Alexander, C. TLR2—Promiscuous or Specific? A Critical Re-Evaluation of a Receptor Expressing Apparent Broad Specificity. Immunobiology 2008, 213, 205–224. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Girardin, S.E.; Philpott, D.J.; Blanot, D.; Nahori, M.-A.; Werts, C.; Boneca, I.G. Toll-like Receptor 2-Dependent Bacterial Sensing Does Not Occur via Peptidoglycan Recognition. EMBO Rep. 2004, 5, 1000–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Adachi, Y.; Ishii, T.; Miura, N.; Tamura, H.; Ohno, N. Dissociation of Toll-like Receptor 2-Mediated Innate Immune Response to Zymosan by Organic Solvent-Treatment without Loss of Dectin-1 Reactivity. Biol. Pharm. Bull. 2008, 31, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Anstett, M.A.; Müller, P.; Albrecht, T.; Nega, M.; Wagener, J.; Gao, Q.; Kaesler, S.; Schaller, M.; Biedermann, T.; Götz, F. Staphylococcal Peptidoglycan Co-Localizes with Nod2 and TLR2 and Activates Innate Immune Response via Both Receptors in Primary Murine Keratinocytes. PLoS ONE 2010, 5, e13153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbet, M.B.; Sefton, M.V. Endotoxin: The Uninvited Guest. Biomaterials 2005, 26, 6811–6817. [Google Scholar] [CrossRef]
- Pacheco, K.A.; McCammon, C.; Thorne, P.S.; O’Neill, M.E.; Liu, A.H.; Martyny, J.W.; Vandyke, M.; Newman, L.S.; Rose, C.S. Characterization of Endotoxin and Mouse Allergen Exposures in Mouse Facilities and Research Laboratories. Ann. Occup. Hyg. 2006, 50, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Thorne, P.S.; Mendy, A.; Metwali, N.; Salo, P.; Co, C.; Jaramillo, R.; Rose, K.M.; Zeldin, D.C. Endotoxin Exposure: Predictors and Prevalence of Associated Asthma Outcomes in the United States. Am. J. Respir. Crit. Care Med. 2015, 192, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Tsan, M.-F.; Gao, B. Pathogen-Associated Molecular Pattern Contamination as Putative Endogenous Ligands of Toll-like Receptors. J. Endotoxin. Res. 2007, 13, 6–14. [Google Scholar] [CrossRef]
- Morrison, D.C.; Jacobs, D.M. Binding of Polymyxin B to the Lipid A Portion of Bacterial Lipopolysaccharides. Immunochemistry 1976, 13, 813–818. [Google Scholar] [CrossRef]
- Akashi, S.; Nagai, Y.; Ogata, H.; Oikawa, M.; Fukase, K.; Kusumoto, S.; Kawasaki, K.; Nishijima, M.; Hayashi, S.; Kimoto, M.; et al. Human MD-2 Confers on Mouse Toll-like Receptor 4 Species-Specific Lipopolysaccharide Recognition. Int. Immunol. 2001, 13, 1595–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjar, A.M.; Tsai, J.H.; Wilson, C.B.; Miller, S.I. Human Toll-like Receptor 4 Recognizes Host-Specific LPS Modifications. Nat. Immunol. 2002, 3, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.I.; Akashi, S.; Yamada, T.; Tanimura, N.; Kobayashi, M.; Konno, K.; Matsumoto, F.; Fukase, K.; Kusumoto, S.; Nagai, Y.; et al. Lipid A Antagonist, Lipid IVa, Is Distinct from Lipid A in Interaction with Toll-like Receptor 4 (TLR4)-MD-2 and Ligand-Induced TLR4 Oligomerization. Int. Immunol. 2004, 16, 961–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, S.A.; Kagan, J.C. Cytosolic Detection of Phagosomal Bacteria-Mechanisms Underlying PAMP Exodus from the Phagosome into the Cytosol. Mol. Microbiol. 2021, 116, 1420–1432. [Google Scholar] [CrossRef]
- Margine, I.; Martinez-Gil, L.; Chou, Y.-Y.; Krammer, F. Residual Baculovirus in Insect Cell-Derived Influenza Virus-like Particle Preparations Enhances Immunogenicity. PLoS ONE 2012, 7, e51559. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.R.; Byregowda, S.M.; Veeregowda, B.M.; Balamurugan, V. An Overview of Heterologous Expression Host Systems for the Production of Recombinant Proteins. Adv. Anim. Vet. Sci. 2016, 4, 346–356. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatton, A.A.; Guerra, F.E. Scratching the Surface Takes a Toll: Immune Recognition of Viral Proteins by Surface Toll-like Receptors. Viruses 2023, 15, 52. https://doi.org/10.3390/v15010052
Hatton AA, Guerra FE. Scratching the Surface Takes a Toll: Immune Recognition of Viral Proteins by Surface Toll-like Receptors. Viruses. 2023; 15(1):52. https://doi.org/10.3390/v15010052
Chicago/Turabian StyleHatton, Alexis A., and Fermin E. Guerra. 2023. "Scratching the Surface Takes a Toll: Immune Recognition of Viral Proteins by Surface Toll-like Receptors" Viruses 15, no. 1: 52. https://doi.org/10.3390/v15010052
APA StyleHatton, A. A., & Guerra, F. E. (2023). Scratching the Surface Takes a Toll: Immune Recognition of Viral Proteins by Surface Toll-like Receptors. Viruses, 15(1), 52. https://doi.org/10.3390/v15010052