
Long-Read High-Throughput Sequencing (HTS) Revealed That the Sf-Rhabdovirus X+ Genome Contains a 3.7 kb Internal Duplication


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sf9 Cells and Sf9 Cell Clones
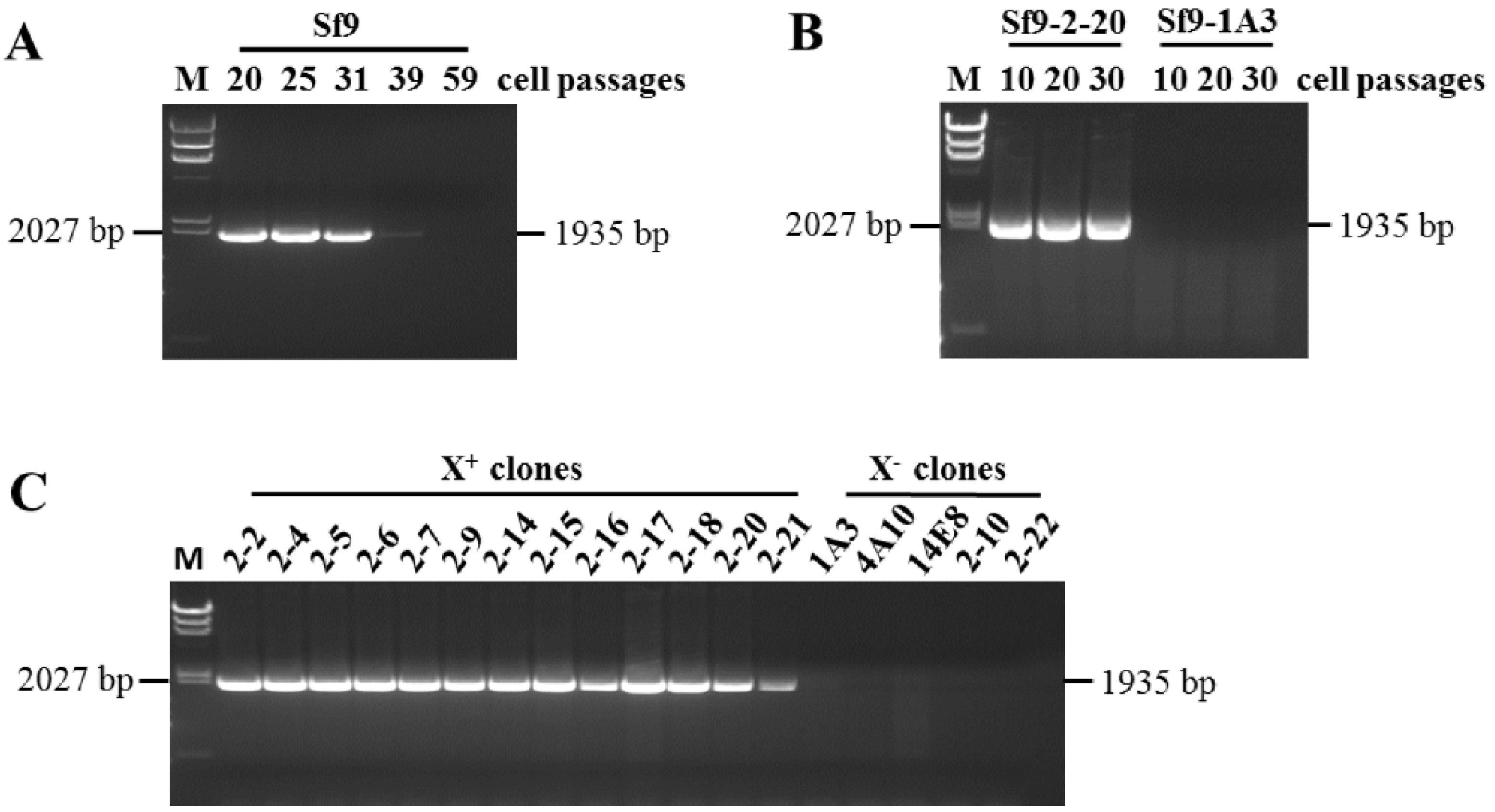
2.2. RT-PCR Analysis
2.3. PacBio HTS Sequencing
2.4. Oxford Nanopore HTS Sequencing
2.5. Illumina HiSeq HTS Sequencing
3. Results
3.1. Identification of X+3.7 in Sf9 Cell Transcriptome
3.2. Identification of X+3.7 Produced by Sf9 X+ Cell Clones
3.3. Oxford Nanopore Sequencing of the X+3.7 Virus Produced from Sf-2-20 Cell Clone
3.4. Characterization of X+3.7 Genome Structure
3.5. Analysis of HTS Long-Reads and Short-Reads for Assembly of Sf- Rhabdovirus X+3.7 Genome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zanella, M.C.; Cordey, S.; Laubscher, F.; Docquier, M.; Vieille, G.; Van Delden, C.; Braunersreuther, V.; Ta, M.K.; Lobrinus, J.A.; Masouridi-Levrat, S.; et al. Unmasking viral sequences by metagenomic next-generation sequencing in adult human blood samples during steroid-refractory/dependent graft-versus-host disease. Microbiome 2021, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Kim, W.K.; No, J.S.; Lee, S.H.; Jung, J.; Yi, Y.; Park, H.C.; Lee, G.Y.; Park, K.; Kim, J.A.; et al. Urinary genome detection and tracking of Hantaan virus from hemorrhagic fever with renal syndrome patients using multiplex PCR-based next-generation sequencing. PLoS Negl. Trop. Dis. 2021, 15, e0009707. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, G.; Desfarges, S.; Uyttebroeck, F.; Munoz, M.; Beerenwinkel, N.; Rougemont, J.; Telenti, A.; Ciuffi, A. Analysis of HIV-1 expression level and sense of transcription by high-throughput sequencing of the infected cell. J. Virol. 2011, 85, 6205–6211. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, S.D.; Narayan, R.; Latham, S.; Lee, S.; Antrobus, R.; Gangadharan, B.; Luo, S.; Schroth, G.P.; Klenerman, P.; Zitzmann, N. Transcriptome sequencing, microarray, and proteomic analyses reveal cellular and metabolic impact of hepatitis C virus infection in vitro. Hepatology 2010, 52, 443–453. [Google Scholar] [CrossRef]
- Amit, I.; Garber, M.; Chevrier, N.; Leite, A.P.; Donner, Y.; Eisenhaure, T.; Guttman, M.; Grenier, J.K.; Li, W.; Zuk, O.; et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 2009, 326, 257–263. [Google Scholar] [CrossRef]
- Grimaldi, A.; Panariello, F.; Annunziata, P.; Giuliano, T.; Daniele, M.; Pierri, B.; Colantuono, C.; Salvi, M.; Bouche, V.; Manfredi, A.; et al. Improved SARS-CoV-2 sequencing surveillance allows the identification of new variants and signatures in infected patients. Genome Med. 2022, 14, 90. [Google Scholar] [CrossRef]
- Quer, J.; Colomer-Castell, S.; Campos, C.; Andres, C.; Pinana, M.; Cortese, M.F.; Gonzalez-Sanchez, A.; Garcia-Cehic, D.; Ibanez, M.; Pumarola, T.; et al. Next-Generation Sequencing for Confronting Virus Pandemics. Viruses 2022, 14, 600. [Google Scholar] [CrossRef]
- Neverov, A.; Chumakov, K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc. Natl. Acad. Sci. USA 2010, 107, 20063–20068. [Google Scholar] [CrossRef]
- Ma, H.; Ma, Y.; Ma, W.; Williams, D.K.; Galvin, T.A.; Khan, A.S. Chemical induction of endogenous retrovirus particles from the vero cell line of African green monkeys. J. Virol. 2011, 85, 6579–6588. [Google Scholar] [CrossRef]
- Onions, D.; Cote, C.; Love, B.; Toms, B.; Koduri, S.; Armstrong, A.; Chang, A.; Kolman, J. Ensuring the safety of vaccine cell substrates by massively parallel sequencing of the transcriptome. Vaccine 2011, 29, 7117–7121. [Google Scholar] [CrossRef]
- Ma, H.; Galvin, T.A.; Glasner, D.R.; Shaheduzzaman, S.; Khan, A.S. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J. Virol. 2014, 88, 6576–6585. [Google Scholar] [CrossRef] [PubMed]
- Victoria, J.G.; Wang, C.; Jones, M.S.; Jaing, C.; McLoughlin, K.; Gardner, S.; Delwart, E.L. Viral nucleic acids in live-attenuated vaccines: Detection of minority variants and an adventitious virus. J. Virol. 2010, 84, 6033–6040. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Nandakumar, S.; Bae, E.H.; Chin, P.J.; Khan, A.S. The Spodoptera frugiperda Sf9 cell line is a heterogeneous population of rhabdovirus-infected and virus-negative cells: Isolation and characterization of cell clones containing rhabdovirus X-gene variants and virus-negative cell clones. Virology 2019, 536, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Maghodia, A.B.; Jarvis, D.L. Infectivity of Sf-rhabdovirus variants in insect and mammalian cell lines. Virology 2017, 512, 234–245. [Google Scholar] [CrossRef]
- Schroeder, L.; Mar, T.B.; Haynes, J.R.; Wang, R.; Wempe, L.; Goodin, M.M. Host Range and Population Survey of Spodoptera frugiperda Rhabdovirus. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Macri, D.; Srivastava, I.; McPherson, C.; Felberbaum, R.; Post, P.; Cox, M. Complete study demonstrating the absence of rhabdovirus in a distinct Sf9 cell line. PLoS ONE 2017, 12, e0175633. [Google Scholar] [CrossRef]
- Deschamps-Francoeur, G.; Simoneau, J.; Scott, M.S. Handling multi-mapped reads in RNA-seq. Comput. Struct. Biotechnol. J. 2020, 18, 1569–1576. [Google Scholar] [CrossRef]
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of genome size and complexity in the rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef]
- Blasdell, K.R.; Widen, S.G.; Diviney, S.M.; Firth, C.; Wood, T.G.; Guzman, H.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N.; Walker, P.J. Koolpinyah and Yata viruses: Two newly recognised ephemeroviruses from tropical regions of Australia and Africa. Vet. Microbiol. 2014, 174, 547–553. [Google Scholar] [CrossRef]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus accessory genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef]
- Walker, P.J.; Byrne, K.A.; Riding, G.A.; Cowley, J.A.; Wang, Y.; McWilliam, S. The genome of bovine ephemeral fever rhabdovirus contains two related glycoprotein genes. Virology 1992, 191, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.B.; Mead, D.G.; Palacios, G.F.; Tesh, R.B.; Holmes, E.C. Gene duplication and phylogeography of North American members of the Hart Park serogroup of avian rhabdoviruses. Virology 2014, 448, 284–292. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample IDs | Total No. of Reads | No. of Reads Past Q7 Filter | Average Q7 Past Read Length (bp) | No. of Rhabdovirus Reads (CLC) | Average Rhabdovirus Read Length (bp) |
|---|---|---|---|---|---|
| 2B | 28,062 | 5679 | 831 | 562 | 2481 |
| 2A | 233,281 | 93,287 | 570 | 92 | 2744 |
| 1 | 571,301 | 193,530 | 509 | 229 | 2025 |
| Combined | 292,496 | 883 |
| Read Length (bp) | Aligned Region in KF947078 3→5′ | Identity (%) 1 | Gaps (%) 1 |
|---|---|---|---|
| 16,689 | 659–8523^GA^4767–13,526 (338) 2 | 92.1 | 5.5 |
| 12,529-R 3 | (83)-8523^GA^4767–8673 | 93.0 | 4.9 |
| 11,631 | 353–8523^GA^4767–8295 | 93.5 | 4.6 |
| 11,247-R | (84)-8523^GA^4767–7555 | 91.9 | 5.6 |
| 9135 | (65)-8523^GA^4767–5650 | 88.2 | 8.0 |
| 4634-R | 4407–8523^GA^4767–5375 | 91.1 | 6.5 |
| 4537 | 7054–8523^GA^4767–7613 | 91.4 | 6.2 |
| 2361 | 6297–8523^GA^4767–4895 | 88.6 | 7.2 |
| 916-R | 8096–8523^GA^4767–5275 | 92.8 | 5.7 |
| Putative Genes | Location in KF947078 (nt) | Location in X+3.7 (nt) |
|---|---|---|
| N | 81–1646 | 90–1718 |
| P | 1726–2871 | 1798–2943 |
| M | 3088–4014 | 3160–4086 |
| G | 4140–5972 | 4212–6044 |
| X | 6161–6490 | 6233–6562 |
| L1 | 6833–7074 | 6905–7153 |
| L2 | 7211–7740 | 7290–7817 |
| L3/G1 | 8505–8523^GA^4767–5972 | 8582–9808 |
| X1 | 6161–6490 | 9997–10,326 |
| L | 6833–13,255 | 10,669–17,091 |
| Reference X+ Genome | X+3.7 Genome | ||||
|---|---|---|---|---|---|
| Primary Sequence | Duplication Sequence | ||||
| Location (ORF) | nt | Location (ORF) | nt | Location (ORF) | nt |
| 4878 (G) | T | 4950 (G) | T | 8714 (L3/G1) | C |
| 4905 (G) | C | 4977 (G) | C | 8741 (L3/G1) | T |
| 4908 (G) | G | 4980 (G) | G | 8744 (L3/G1) | A |
| 5003 (G) | G | 5075 (G) | G | 8839 (L3/G1) | A |
| 5036 (G) | G | 5108 (G) | G | 8872 (L3/G1) | A |
| 5578 (G) | G | 5650 (G) | G | 9414 (L3/G1) | A |
| 5687 (G) | G | 5759 (G) | G | 9523 (L3/G1) | A |
| 5817 (G) | A | 5889 (G) | A | 9653 (L3/G1) | G |
| 6333 (X) | C | 6405 (X) | C | 10,169 (X1) | A |
| 6354 (X) | A | 6426 (X) | A | 10,190 (X1) | T |
| 6732 (X-L) | C | 6804 (X-L) | T | 10,568 (X1-L) | C |
| 7016^7017 (L) | - | 7089–7094 (L1) | AAAACC | 10,852^10,853 (L) | - |
| 7018 (L) | A | 7096 (L1) | C | 10,854 (L) | A |
| 7050^7051 (L) | - | 7129 (L1) | G | 10,886^10,887 (L) | - |
| 7452 (L) | C | 7531 (L2) | T | 11,288 (L) | C |
| 7624 (L) | T | 7703 (L2) | G | 11,460 (L) | T |
| 7699–7700 (L) | AG | 7777^7778 (L2) | -- | 11,535–11,536 (L) | AG |
| 8052 (L) | C | 8129 (L2-L3/G1) | T | 11,888 (L) | C |
| 8070 (L) | G | 8147 (L2-L3/G1) | A | 11,906 (L) | G |
| 8104 (L) | G | 8181 (L2-L3/G1) | T | 11,940 (L) | G |
| 8164 (L) | C | 8241 (L2-L3/G1) | T | 12,000 (L) | C |
| 8189 (L) | G | 8266 (L2-L3/G1) | A | 12,025 (L) | G |
| 8351 (L) | G | 8428 (L2-L3/G1) | A | 12,187 (L) | G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, H.; Bosma, T.J.; Khan, A.S. Long-Read High-Throughput Sequencing (HTS) Revealed That the Sf-Rhabdovirus X+ Genome Contains a 3.7 kb Internal Duplication. Viruses 2023, 15, 1998. https://doi.org/10.3390/v15101998
Ma H, Bosma TJ, Khan AS. Long-Read High-Throughput Sequencing (HTS) Revealed That the Sf-Rhabdovirus X+ Genome Contains a 3.7 kb Internal Duplication. Viruses. 2023; 15(10):1998. https://doi.org/10.3390/v15101998
Chicago/Turabian StyleMa, Hailun, Trent J. Bosma, and Arifa S. Khan. 2023. "Long-Read High-Throughput Sequencing (HTS) Revealed That the Sf-Rhabdovirus X+ Genome Contains a 3.7 kb Internal Duplication" Viruses 15, no. 10: 1998. https://doi.org/10.3390/v15101998
APA StyleMa, H., Bosma, T. J., & Khan, A. S. (2023). Long-Read High-Throughput Sequencing (HTS) Revealed That the Sf-Rhabdovirus X+ Genome Contains a 3.7 kb Internal Duplication. Viruses, 15(10), 1998. https://doi.org/10.3390/v15101998