

Host Responses to Respiratory Syncytial Virus Infection

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Innate Immune Responses to RSV Infection
2.1. Infection of Epithelial Cells
2.2. Eosinophils
2.3. Neutrophils
2.4. Natural Killer Cells
2.5. Monocytes
2.6. Macrophages
2.7. Dendritic Cells
3. Adaptive Immune Responses to RSV Infection
3.1. B Cells
3.2. T Cells
3.2.1. CD4+ T Cells
3.2.2. CD8+ T Cells
3.2.3. Regulatory T Cells
3.2.4. Memory T Cells
4. Vaccine Development and Treatment Options with Novel Insights into Immune-Mediated Protection
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory syncytial virus: Virology, reverse genetics, and pathogenesis of disease. Curr. Top. Microbiol. Immunol. 2013, 372, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Mufson, M.A.; Orvell, C.; Rafnar, B.; Norrby, E. Two distinct subtypes of human respiratory syncytial virus. J. Gen. Virol. 1985, 66, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.M.; Fu, Y.H.; Peng, X.L.; Zheng, Y.P.; He, J.S. Genetic diversity and molecular evolution of human respiratory syncytial virus A and B. Sci. Rep. 2021, 11, 12941. [Google Scholar] [CrossRef] [PubMed]
- Cantu-Flores, K.; Rivera-Alfaro, G.; Munoz-Escalante, J.C.; Noyola, D.E. Global distribution of respiratory syncytial virus A and B infections: A systematic review. Pathog. Glob. Health 2022, 116, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory Syncytial Virus Infection in Elderly and High-Risk Adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in elderly adults. Drugs Aging 2005, 22, 577–587. [Google Scholar] [CrossRef]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simoes, E.A.F.; Campbell, H.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis. Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef]
- Savic, M.; Penders, Y.; Shi, T.; Branche, A.; Pircon, J.Y. Respiratory syncytial virus disease burden in adults aged 60 years and older in high-income countries: A systematic literature review and meta-analysis. Influenza Other Respir. Viruses 2023, 17, e13031. [Google Scholar] [CrossRef]
- Chow, E.J.; Uyeki, T.M.; Chu, H.Y. The effects of the COVID-19 pandemic on community respiratory virus activity. Nat. Rev. Microbiol. 2023, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Lin, K.P.; Wang, L.A.; Yeh, T.K.; Liu, P.Y. The Impact of the COVID-19 Pandemic on Respiratory Syncytial Virus Infection: A Narrative Review. Infect. Drug Resist. 2023, 16, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Di Mattia, G.; Nenna, R.; Mancino, E.; Rizzo, V.; Pierangeli, A.; Villani, A.; Midulla, F. During the COVID-19 pandemic where has respiratory syncytial virus gone? Pediatr. Pulmonol. 2021, 56, 3106–3109. [Google Scholar] [CrossRef]
- Smyth, R.L.; Openshaw, P.J. Bronchiolitis. Lancet 2006, 368, 312–322. [Google Scholar] [CrossRef]
- Henderson, F.W.; Collier, A.M.; Clyde, W.A., Jr.; Denny, F.W. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N. Engl. J. Med. 1979, 300, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Glezen, W.P.; Taber, L.H.; Frank, A.L.; Kasel, J.A. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 1986, 140, 543–546. [Google Scholar] [CrossRef]
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef]
- Pickles, R.J.; DeVincenzo, J.P. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J. Pathol. 2015, 235, 266–276. [Google Scholar] [CrossRef]
- McNamara, P.S.; Smyth, R.L. The pathogenesis of respiratory syncytial virus disease in childhood. Br. Med. Bull. 2002, 61, 13–28. [Google Scholar] [CrossRef]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef]
- Walsh, E.E.; Falsey, A.R. Respiratory syncytial virus infection in adult populations. Infect. Disord. Drug Targets 2012, 12, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Ooi, Y.; Zaw, E.M.; Utjesanovic, N.; Campbell, H.; Cunningham, S.; Bont, L.; Nair, H.; Investigators, R. Association Between Respiratory Syncytial Virus-Associated Acute Lower Respiratory Infection in Early Life and Recurrent Wheeze and Asthma in Later Childhood. J. Infect. Dis. 2020, 222, S628–S633. [Google Scholar] [CrossRef]
- Coutts, J.; Fullarton, J.; Morris, C.; Grubb, E.; Buchan, S.; Rodgers-Gray, B.; Thwaites, R. Association between respiratory syncytial virus hospitalization in infancy and childhood asthma. Pediatr. Pulmonol. 2020, 55, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- van Wijhe, M.; Johannesen, C.K.; Simonsen, L.; Jorgensen, I.M.; Investigators, R.; Fischer, T.K. A Retrospective Cohort Study on Infant Respiratory Tract Infection Hospitalizations and Recurrent Wheeze and Asthma Risk: Impact of Respiratory Syncytial Virus. J. Infect. Dis. 2022, 226, S55–S62. [Google Scholar] [CrossRef] [PubMed]
- McGinley, J.P.; Lin, G.L.; Oner, D.; Golubchik, T.; O’Connor, D.; Snape, M.D.; Gruselle, O.; Langedijk, A.C.; Wildenbeest, J.; Openshaw, P.; et al. Clinical and Viral Factors Associated With Disease Severity and Subsequent Wheezing in Infants With Respiratory Syncytial Virus Infection. J. Infect. Dis. 2022, 226, S45–S54. [Google Scholar] [CrossRef]
- Rosas-Salazar, C.; Chirkova, T.; Gebretsadik, T.; Chappell, J.D.; Peebles, R.S., Jr.; Dupont, W.D.; Jadhao, S.J.; Gergen, P.J.; Anderson, L.J.; Hartert, T.V. Respiratory syncytial virus infection during infancy and asthma during childhood in the USA (INSPIRE): A population-based, prospective birth cohort study. Lancet 2023, 401, 1669–1680. [Google Scholar] [CrossRef]
- Glezen, W.P.; Greenberg, S.B.; Atmar, R.L.; Piedra, P.A.; Couch, R.B. Impact of respiratory virus infections on persons with chronic underlying conditions. JAMA 2000, 283, 499–505. [Google Scholar] [CrossRef]
- Fixler, D.E. Respiratory syncytial virus infection in children with congenital heart disease: A review. Pediatr. Cardiol. 1996, 17, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Canchola, J.G.; Brandt, C.D.; Pyles, G.; Chanock, R.M.; Jensen, K.; Parrott, R.H. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 1969, 89, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Pedrero, M.A.; Osterhaus, A.; Becker, T.; Elbahesh, H.; Rimmelzwaan, G.F.; Saletti, G. Aging and Options to Halt Declining Immunity to Virus Infections. Front. Immunol. 2021, 12, 681449. [Google Scholar] [CrossRef]
- Andrade, C.A.; Pacheco, G.A.; Galvez, N.M.S.; Soto, J.A.; Bueno, S.M.; Kalergis, A.M. Innate Immune Components that Regulate the Pathogenesis and Resolution of hRSV and hMPV Infections. Viruses 2020, 12, 637. [Google Scholar] [CrossRef] [PubMed]
- Ballegeer, M.; Saelens, X. Cell-Mediated Responses to Human Metapneumovirus Infection. Viruses 2020, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Farrag, M.A.; Almajhdi, F.N. Human Respiratory Syncytial Virus: Role of Innate Immunity in Clearance and Disease Progression. Viral Immunol. 2016, 29, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lopez, C.B. The innate immune response to RSV: Advances in our understanding of critical viral and host factors. Vaccine 2017, 35, 481–488. [Google Scholar] [CrossRef]
- Turvey, S.E.; Broide, D.H. Innate immunity. J. Allergy Clin. Immunol. 2010, 125, S24–S32. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Openshaw, P.J.M.; Chiu, C.; Culley, F.J.; Johansson, C. Protective and Harmful Immunity to RSV Infection. Annu. Rev. Immunol. 2017, 35, 501–532. [Google Scholar] [CrossRef]
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The airway epithelium: Soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 2011, 24, 210–229. [Google Scholar] [CrossRef]
- Kuek, L.E.; Lee, R.J. First contact: The role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L603–L619. [Google Scholar] [CrossRef]
- Wallace, L.E.; Liu, M.; van Kuppeveld, F.J.M.; de Vries, E.; de Haan, C.A.M. Respiratory mucus as a virus-host range determinant. Trends Microbiol. 2021, 29, 983–992. [Google Scholar] [CrossRef]
- Ridley, C.; Thornton, D.J. Mucins: The frontline defence of the lung. Biochem. Soc. Trans. 2018, 46, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; van Putten, J.P.M.; Strijbis, K. Defensive Properties of Mucin Glycoproteins during Respiratory Infections-Relevance for SARS-CoV-2. mBio 2020, 11, e02374-20. [Google Scholar] [CrossRef] [PubMed]
- LeMessurier, K.S.; Tiwary, M.; Morin, N.P.; Samarasinghe, A.E. Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.J.; Doherty, P.C.; Thomas, P.G. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res. 2011, 343, 13–21. [Google Scholar] [CrossRef]
- Zanin, M.; Baviskar, P.; Webster, R.; Webby, R. The Interaction between Respiratory Pathogens and Mucus. Cell Host Microbe 2016, 19, 159–168. [Google Scholar] [CrossRef]
- Bailey, K.L. Aging Diminishes Mucociliary Clearance of the Lung. Adv. Geriatr. Med. Res. 2022, 4, e220005. [Google Scholar] [CrossRef]
- Ioannidis, I.; McNally, B.; Willette, M.; Peeples, M.E.; Chaussabel, D.; Durbin, J.E.; Ramilo, O.; Mejias, A.; Flano, E. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J. Virol. 2012, 86, 5422–5436. [Google Scholar] [CrossRef]
- Zhang, L.; Peeples, M.E.; Boucher, R.C.; Collins, P.L.; Pickles, R.J. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 2002, 76, 5654–5666. [Google Scholar] [CrossRef]
- Barnes, M.V.C.; Openshaw, P.J.M.; Thwaites, R.S. Mucosal Immune Responses to Respiratory Syncytial Virus. Cells 2022, 11, 1153. [Google Scholar] [CrossRef]
- Feng, Z.; Xu, L.; Xie, Z. Receptors for Respiratory Syncytial Virus Infection and Host Factors Regulating the Life Cycle of Respiratory Syncytial Virus. Front. Cell Infect. Microbiol. 2022, 12, 858629. [Google Scholar] [CrossRef]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, P.; Chin, A.A.; Tan, S.; Jeon, A.H.; Ackerley, C.A.; Siu, K.K.; Lee, J.E.; Hegele, R.G. Identification of RSV Fusion Protein Interaction Domains on the Virus Receptor, Nucleolin. Viruses 2021, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Green, G.; Johnson, S.M.; Costello, H.; Brakel, K.; Harder, O.; Oomens, A.G.; Peeples, M.E.; Moulton, H.M.; Niewiesk, S. CX3CR1 Is a Receptor for Human Respiratory Syncytial Virus in Cotton Rats. J. Virol. 2021, 95, e0001021. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; McNally, B.A.; Ioannidis, I.; Flano, E.; Teng, M.N.; Oomens, A.G.; Walsh, E.E.; Peeples, M.E. Respiratory Syncytial Virus Uses CX3CR1 as a Receptor on Primary Human Airway Epithelial Cultures. PLoS Pathog. 2015, 11, e1005318. [Google Scholar] [CrossRef]
- Chirkova, T.; Lin, S.; Oomens, A.G.P.; Gaston, K.A.; Boyoglu-Barnum, S.; Meng, J.; Stobart, C.C.; Cotton, C.U.; Hartert, T.V.; Moore, M.L.; et al. CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J. Gen. Virol. 2015, 96, 2543–2556. [Google Scholar] [CrossRef]
- Anderson, C.S.; Chu, C.Y.; Wang, Q.; Mereness, J.A.; Ren, Y.; Donlon, K.; Bhattacharya, S.; Misra, R.S.; Walsh, E.E.; Pryhuber, G.S.; et al. CX3CR1 as a respiratory syncytial virus receptor in pediatric human lung. Pediatr. Res. 2020, 87, 862–867. [Google Scholar] [CrossRef]
- Krusat, T.; Streckert, H.J. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 1997, 142, 1247–1254. [Google Scholar] [CrossRef]
- Bourgeois, C.; Bour, J.B.; Lidholt, K.; Gauthray, C.; Pothier, P. Heparin-like structures on respiratory syncytial virus are involved in its infectivity in vitro. J. Virol. 1998, 72, 7221–7227. [Google Scholar] [CrossRef]
- Feldman, S.A.; Audet, S.; Beeler, J.A. The Fusion Glycoprotein of Human Respiratory Syncytial Virus Facilitates Virus Attachment and Infectivity via an Interaction with Cellular Heparan Sulfate. J. Virol. 2000, 74, 6442–6447. [Google Scholar] [CrossRef]
- Behera, A.K.; Matsuse, H.; Kumar, M.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Blocking intercellular adhesion molecule-1 on human epithelial cells decreases respiratory syncytial virus infection. Biochem. Biophys. Res. Commun. 2001, 280, 188–195. [Google Scholar] [CrossRef]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Vallon-Eberhard, A.; Zigmond, E.; Farache, J.; Shezen, E.; Shakhar, G.; Ludwig, A.; Lira, S.A.; Jung, S. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 2011, 118, e156–e167. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef]
- Tripp, R.A.; Jones, L.P.; Haynes, L.M.; Zheng, H.; Murphy, P.M.; Anderson, L.J. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat. Immunol. 2001, 2, 732–738. [Google Scholar] [CrossRef]
- Ha, B.; Chirkova, T.; Boukhvalova, M.S.; Sun, H.Y.; Walsh, E.E.; Anderson, C.S.; Mariani, T.J.; Anderson, L.J. Mutation of Respiratory Syncytial Virus G Protein’s CX3C Motif Attenuates Infection in Cotton Rats and Primary Human Airway Epithelial Cells. Vaccines 2019, 7, 69. [Google Scholar] [CrossRef]
- Malik, G.; Zhou, Y. Innate Immune Sensing of Influenza A Virus. Viruses 2020, 12, 755. [Google Scholar] [CrossRef]
- Islamuddin, M.; Mustfa, S.A.; Ullah, S.; Omer, U.; Kato, K.; Parveen, S. Innate Immune Response and Inflammasome Activation During SARS-CoV-2 Infection. Inflammation 2022, 45, 1849–1863. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Sen, G.C. Innate Immune Responses to Herpesvirus Infection. Cells 2021, 10, 2122. [Google Scholar] [CrossRef]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef]
- Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11, 171. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Rudd, B.D.; Burstein, E.; Duckett, C.S.; Li, X.; Lukacs, N.W. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Virol. 2005, 79, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Bueno, S.M.; Gonzalez, P.A.; Riedel, C.A.; Carreno, L.J.; Vasquez, A.E.; Kalergis, A.M. Local cytokine response upon respiratory syncytial virus infection. Immunol. Lett. 2011, 136, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Noah, T.L.; Becker, S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am. J. Physiol. 1993, 265, 472–478. [Google Scholar] [CrossRef]
- Noah, T.L.; Henderson, F.W.; Wortman, I.A.; Devlin, R.B.; Handy, J.; Koren, H.S.; Becker, S. Nasal cytokine production in viral acute upper respiratory infection of childhood. J. Infect. Dis. 1995, 171, 584–592. [Google Scholar] [CrossRef]
- Saito, T.; Deskin, R.W.; Casola, A.; Häeberle, H.; Olszewska, B.; Ernst, P.B.; Alam, R.; Ogra, P.L.; Garofalo, R. Respiratory Syncytial Virus Induces Selective Production of the Chemokine RANTES by Upper Airway Epithelial Cells. J. Infect. Dis. 1996, 175, 497–504. [Google Scholar] [CrossRef]
- Olszewska-Pazdrak, B.; Casola, A.; Saito, T.; Alam, R.; Crowe, S.E.; Mei, F.; Ogra, P.L.; Garofalo, R.P. Cell-Specific Expression of RANTES, MCP-1, and MIP-1a by Lower Airway Epithelial Cells and Eosinophils Infected with Respiratory Syncytial Virus. J. Virol. 1998, 72, 4756–4764. [Google Scholar] [CrossRef]
- Wang, S.-Z.; Hallsworth, P.G.; Dowling, K.D.; Alpers, J.H.; Bowden, J.J.; Forsyth, K.D. Adhesion molecule expression on epithelial cells infected with respiratory syncytial virus. Eur. Respir. J. 2000, 15, 358–366. [Google Scholar] [CrossRef]
- Guo, X.; Liu, T.; Shi, H.; Wang, J.; Ji, P.; Wang, H.; Hou, Y.; Tan, R.X.; Li, E. Respiratory Syncytial Virus Infection Upregulates NLRC5 and Major Histocompatibility Complex Class I Expression through RIG-I Induction in Airway Epithelial Cells. J. Virol. 2015, 89, 7636–7645. [Google Scholar] [CrossRef]
- Levitz, R.; Wattier, R.; Phillips, P.; Solomon, A.; Lawler, J.; Lazar, I.; Weibel, C.; Kahn, J.S. Induction of IL-6 and CCL5 (RANTES) in human respiratory epithelial (A549) cells by clinical isolates of respiratory syncytial virus is strain specific. Virol. J. 2012, 9, 190. [Google Scholar] [CrossRef]
- Touzelet, O.; Broadbent, L.; Armstrong, S.D.; Aljabr, W.; Cloutman-Green, E.; Power, U.F.; Hiscox, J.A. The Secretome Profiling of a Pediatric Airway Epithelium Infected with hRSV Identified Aberrant Apical/Basolateral Trafficking and Novel Immune Modulating (CXCL6, CXCL16, CSF3) and Antiviral (CEACAM1) Proteins. Mol. Cell Proteom. 2020, 19, 793–807. [Google Scholar] [CrossRef]
- Van Royen, T.; Rossey, I.; Sedeyn, K.; Schepens, B.; Saelens, X. How RSV Proteins Join Forces to Overcome the Host Innate Immune Response. Viruses 2022, 14, 419. [Google Scholar] [CrossRef]
- Moore, E.C.; Barber, J.; Tripp, R.A. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol. J. 2008, 5, 116. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, J.; Zheng, K.; Hou, Y.; Zhao, F.; Zhao, D. Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology 2014, 57, 65–73. [Google Scholar] [CrossRef]
- Pei, J.; Beri, N.R.; Zou, A.J.; Hubel, P.; Dorando, H.K.; Bergant, V.; Andrews, R.D.; Pan, J.; Andrews, J.M.; Sheehan, K.C.F.; et al. Nuclear-localized human respiratory syncytial virus NS1 protein modulates host gene transcription. Cell Rep. 2021, 37, 109803. [Google Scholar] [CrossRef]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef]
- Bitko, V.; Shulyayeva, O.; Mazumder, B.; Musiyenko, A.; Ramaswamy, M.; Look, D.C.; Barik, S. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 2007, 81, 1786–1795. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tsutsumi, H.; Osaki, M.; Haseyama, K.; Mizue, N.; Chiba, C. Respiratory syncytial virus infection of human alveolar epithelial cells enhances interferon regulatory factor 1 and interleukin-1beta-converting enzyme gene expression but does not cause apoptosis. J. Virol. 1998, 72, 4498–4502. [Google Scholar] [CrossRef]
- Simpson, J.; Loh, Z.; Ullah, M.A.; Lynch, J.P.; Werder, R.B.; Collinson, N.; Zhang, V.; Dondelinger, Y.; Bertrand, M.J.M.; Everard, M.L.; et al. Respiratory Syncytial Virus Infection Promotes Necroptosis and HMGB1 Release by Airway Epithelial Cells. Am. J. Respir. Crit. Care Med. 2020, 201, 1358–1371. [Google Scholar] [CrossRef]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E., Jr.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.F.; Rameix-Welti, M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 563. [Google Scholar] [CrossRef]
- Groskreutz, D.J.; Babor, E.C.; Monick, M.M.; Varga, S.M.; Hunninghake, G.W. Respiratory syncytial virus limits alpha subunit of eukaryotic translation initiation factor 2 (eIF2alpha) phosphorylation to maintain translation and viral replication. J. Biol. Chem. 2010, 285, 24023–24031. [Google Scholar] [CrossRef]
- Meineke, R.; Stelz, S.; Busch, M.; Werlein, C.; Kühnel, M.; Jonigk, D.; Rimmelzwaan, G.F.; Elbahesh, H. FDA-approved Abl/EGFR/PDGFR kinase inhibitors show potent efficacy against pandemic and seasonal influenza A virus infections of human lung explants. iScience 2023, 26, 106309. [Google Scholar] [CrossRef]
- Harcourt, J.L.; Haynes, L.M. Establishing a liquid-covered culture of polarized human airway epithelial Calu-3 cells to study host cell response to respiratory pathogens in vitro. J. Vis. Exp. 2013, 72, e50157. [Google Scholar] [CrossRef]
- Ito, K.; Daly, L.; Coates, M. An impact of age on respiratory syncytial virus infection in air-liquid-interface culture bronchial epithelium. Front Med. 2023, 10, 1144050. [Google Scholar] [CrossRef]
- Rajan, A.; Weaver, A.M.; Aloisio, G.M.; Jelinski, J.; Johnson, H.L.; Venable, S.F.; McBride, T.; Aideyan, L.; Piedra, F.A.; Ye, X.; et al. The human nose organoid respiratory virus model: An ex-vivo human challenge model to study RSV and SARS-CoV-2 pathogenesis and evaluate therapeutics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef] [PubMed]
- Flores-Torres, A.S.; Salinas-Carmona, M.C.; Salinas, E.; Rosas-Taraco, A.G. Eosinophils and Respiratory Viruses. Viral Immunol. 2019, 32, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, A.E.; Melo, R.C.; Duan, S.; LeMessurier, K.S.; Liedmann, S.; Surman, S.L.; Lee, J.J.; Hurwitz, J.L.; Thomas, P.G.; McCullers, J.A. Eosinophils Promote Antiviral Immunity in Mice Infected with Influenza A Virus. J. Immunol. 2017, 198, 3214–3226. [Google Scholar] [CrossRef]
- Drake, M.G.; Bivins-Smith, E.R.; Proskocil, B.J.; Nie, Z.; Scott, G.D.; Lee, J.J.; Lee, N.A.; Fryer, A.D.; Jacoby, D.B. Human and Mouse Eosinophils Have Antiviral Activity against Parainfluenza Virus. Am. J. Respir. Cell Mol. Biol. 2016, 55, 387–394. [Google Scholar] [CrossRef]
- Handzel, Z.T.; Busse, W.W.; Sedgwick, J.B.; Vrtis, R.; Lee, W.M.; Kelly, E.A.B.; Gern, J.E. Eosinophils Bind Rhinovirus and Activate Virus-Specific T Cells. J. Immunol. 1998, 160, 1279–1284. [Google Scholar] [CrossRef]
- Ho, K.S.; Howell, D.; Rogers, L.; Narasimhan, B.; Verma, H.; Steiger, D. The relationship between asthma, eosinophilia, and outcomes in coronavirus disease 2019 infection. Ann. Allergy Asthma Immunol. 2021, 127, 42–48. [Google Scholar] [CrossRef]
- Becker, S.; Soukup, J.M. Airway epithelial cell-induced activation of monocytes and eosinophils in respiratory syncytial viral infection. Immunobiology 1999, 201, 88–106. [Google Scholar] [CrossRef]
- Soukup, J.M.; Becker, S. Role of monocytes and eosinophils in human respiratory syncytial virus infection in vitro. Clin. Immunol. 2003, 107, 178–185. [Google Scholar] [CrossRef]
- Harrison, A.M.; Bonville, C.A.; Rosenberg, H.F.; Domachowske, J.B. Respiratory syncytical virus-induced chemokine expression in the lower airways: Eosinophil recruitment and degranulation. Am. J. Respir. Crit. Care Med. 1999, 159, 1918–1924. [Google Scholar] [CrossRef]
- Phipps, S.; Lam, C.E.; Mahalingam, S.; Newhouse, M.; Ramirez, R.; Rosenberg, H.F.; Foster, P.S.; Matthaei, K.I. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood 2007, 110, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Kimpen, J.L.; Luijk, B.; Heidema, J.; Kanters, D.; van der Ent, C.K.; Koenderman, L. Systemic eosinophil response induced by respiratory syncytial virus. Clin. Exp. Immunol. 2006, 144, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.M.; Godding, V.; Sedgwick, J.B.; Busse, W.W. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. Roles of CD18 and intercellular adhesion molecule-1. J. Immunol. 1996, 156, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Olszewska-Pazdrak, B.; Pazdrak, K.; Ogra, P.L.; Garofalo, R.P. Respiratory Syncytial Virus-Infected Pulmonary Epithelial Cells Induce Eosinophil Degranulation by a CD18-Mediated Mechanism. J. Immunol. 1998, 160, 4889–4895. [Google Scholar] [CrossRef]
- Castilow, E.M.; Legge, K.L.; Varga, S.M. Cutting edge: Eosinophils do not contribute to respiratory syncytial virus vaccine-enhanced disease. J. Immunol. 2008, 181, 6692–6696. [Google Scholar] [CrossRef]
- Prince, G.A.; Jenson, A.B.; Hemming, V.; Murphy, B.; Walsh, E.; Horswood, R.; Chanock, R. Enhancement of respiratory syncytial virus pulmonary pathology in cotton rats by prior intramuscular inoculation of formalin-inactiva ted virus. J. Virol. 1986, 57, 721–728. [Google Scholar] [CrossRef]
- Durant, L.R.; Makris, S.; Voorburg, C.M.; Loebbermann, J.; Johansson, C.; Openshaw, P.J. Regulatory T cells prevent Th2 immune responses and pulmonary eosinophilia during respiratory syncytial virus infection in mice. J. Virol. 2013, 87, 10946–10954. [Google Scholar] [CrossRef]
- Conners, M.; Giese, N.A.; Kulkarni, A.B.; Firestone, C.Y.; Morse, H.C.r.; Murphy, B.R. Enhanced pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of formalin-inactivated RSV-immunized BALB/c mice is abrogated by depletion of interleukin-4 (IL-4) and IL-10. J. Virol. 1994, 68, 5321–5325. [Google Scholar] [CrossRef]
- Garofalo, R.; Kimpen, J.L.L.; Welliver, R.C.; Ogra, P.L. Eosinophil degranulation in the respiratory tract during naturally acquired respiratory syncytial virus infection. J. Pediatr. 1992, 120, 28–32. [Google Scholar] [CrossRef]
- Dyer, K.D.; Percopo, C.M.; Fischer, E.R.; Gabryszewski, S.J.; Rosenberg, H.F. Pneumoviruses infect eosinophils and elicit MyD88-dependent release of chemoattractant cytokines and interleukin-6. Blood 2009, 114, 2649–2656. [Google Scholar] [CrossRef]
- Kimpen, J.L.; Garofalo, R.; Welliver, R.C.; Fujihara, K.; Ogra, P.L. An ultrastructural study of the interaction of human eosinophils with respiratory syncytial virus. Pediatr. Allergy Immunol. 1996, 7, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kimpen, J.L.L.; Garofalo, R.; Welliver, R.C.; Ogra, P.L. Activation of Human Eosinophils In vitro by Respiratory Syncytial Virus. Pediatr. Res. 1992, 32, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, J.; Cieslewicz, G.; Hamelmann, E.; Joetham, A.; Shultz, L.D.; Lamers, M.C.; Gelfand, E.W. IL-5 and Eosinophils Are Essential for the Development of Airway Hyperresponsiveness Following Acute Respiratory Syncytial Virus Infection. J. Immunol. 1999, 162, 2997–3004. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, J.; Zhao, Y.; Shan, J.; Wang, L.; Yang, G.; He, S.; Li, E. RSV Infection in Neonatal Mice Induces Pulmonary Eosinophilia Responsible for Asthmatic Reaction. Front. Immunol. 2022, 13, 817113. [Google Scholar] [CrossRef]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Zhu, L. Role of neutrophils in acute viral infection. Immun. Inflamm. Dis. 2021, 9, 1186–1196. [Google Scholar] [CrossRef]
- McNamara, P.S.; Flanagan, B.F.; Hart, C.A.; Smyth, R.L. Production of chemokines in the lungs of infants with severe respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2005, 191, 1225–1232. [Google Scholar] [CrossRef]
- Bataki, E.L.; Evans, G.S.; Everard, M.L. Respiratory syncytial virus and neutrophil activation. Clin. Exp. Immunol. 2005, 140, 470–477. [Google Scholar] [CrossRef]
- Deng, Y.; Herbert, J.A.; Smith, C.M.; Smyth, R.L. An in vitro transepithelial migration assay to evaluate the role of neutrophils in Respiratory Syncytial Virus (RSV) induced epithelial damage. Sci. Rep. 2018, 8, 6777. [Google Scholar] [CrossRef]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.S.S.; Barbosa, L.V.; Diniz, L.F.A.; da Silva, G.S.; Lopes, B.R.P.; Souza, P.M.R.; de Araujo, G.C.; Pessoa, D.; de Oliveira, J.; Souza, F.P.; et al. Neutrophil extracellular traps possess anti-human respiratory syncytial virus activity: Possible interaction with the viral F protein. Virus Res. 2018, 251, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Herbert, J.A.; Robinson, E.; Ren, L.; Smyth, R.L.; Smith, C.M. Neutrophil-Airway Epithelial Interactions Result in Increased Epithelial Damage and Viral Clearance during Respiratory Syncytial Virus Infection. J. Virol. 2020, 94, e02161-19. [Google Scholar] [CrossRef] [PubMed]
- McNamara, P.S.; Ritson, P.; Selby, A.; Hart, C.A.; Smyth, R.L. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 2003, 88, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Everard, M.L.; Swarbrick, A.; Wrightham, M.; McIntyre, J.; Dunkley, C.; James, P.D.; Sewell, H.F.; Milner, A.D. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch. Dis. Child. 1994, 71, 428–432. [Google Scholar] [CrossRef]
- Wang, S.Z.; Xu, H.; Wraith, A.; Bowden, J.J.; Alpers, J.H.; Forsyth, K.D. Neutrophils induce damage to respiratory epithelial cells infected with respiratory syncytial virus. Eur. Respir. J. 1998, 12, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Acosta, P.L.; Caballero, M.T.; Polack, F.P. Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease. Clin. Vaccine Immunol. 2015, 23, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Halfhide, C.P.; Flanagan, B.F.; Brearey, S.P.; Hunt, J.A.; Fonceca, A.M.; McNamara, P.S.; Howarth, D.; Edwards, S.; Smyth, R.L. Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J. Infect. Dis. 2011, 204, 451–458. [Google Scholar] [CrossRef]
- Jaovisidha, P.; Peeples, M.E.; Brees, A.A.; Carpenter, L.R.; Moy, J.N. Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J. Immunol. 1999, 163, 2816–2820. [Google Scholar] [CrossRef]
- Cortjens, B.; de Boer, O.J.; de Jong, R.; Antonis, A.F.; Sabogal Pineros, Y.S.; Lutter, R.; van Woensel, J.B.; Bem, R.A. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef]
- Habibi, M.S.; Thwaites, R.S.; Chang, M.; Jozwik, A.; Paras, A.; Kirsebom, F.; Varese, A.; Owen, A.; Cuthbertson, L.; James, P.; et al. Neutrophilic inflammation in the respiratory mucosa predisposes to RSV infection. Science 2020, 370, eaba9301. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lodoen, M.B.; Lanier, L.L. Natural killer cells as an initial defense against pathogens. Curr. Opin. Immunol. 2006, 18, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Farrag, M.A.; Almajhdi, F.N. Double-edged role of natural killer cells during RSV infection. Int. Rev. Immunol. 2020, 39, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Openshaw, P.J. Intracellular IFN-? expression in natural killer cells precedes lung CD8 T cell recruitment during respiratory syncytial virus infectio. J. Gen. Virol. 1998, 79, 2593–2601. [Google Scholar] [CrossRef]
- Hussell, T.; Openshaw, P.J. IL-12-activated NK cells reduce lung eosinophilia to the attachment protein of respiratory syncytial virus but do not enhance the severity of illness in CD8 T cell-immunodeficient conditions. J. Immunol. 2000, 165, 7109–7115. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Phipps, S.; Angkasekwinai, P.; Dong, C.; Foster, P.S. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J. Immunol. 2010, 185, 4681–4690. [Google Scholar] [CrossRef]
- Welliver, T.P.; Garofalo, R.P.; Hosakote, Y.; Hintz, K.H.; Avendano, L.; Sanchez, K.; Velozo, L.; Jafri, H.; Chavez-Bueno, S.; Ogra, P.L.; et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 2007, 195, 1126–1136. [Google Scholar] [CrossRef]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef]
- Li, F.; Zhu, H.; Sun, R.; Wei, H.; Tian, Z. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J. Virol. 2012, 86, 2251–2258. [Google Scholar] [CrossRef]
- Long, X.; Xie, J.; Zhao, K.; Li, W.; Tang, W.; Chen, S.; Zang, N.; Ren, L.; Deng, Y.; Xie, X.; et al. NK cells contribute to persistent airway inflammation and AHR during the later stage of RSV infection in mice. Med. Microbiol. Immunol. 2016, 205, 459–470. [Google Scholar] [CrossRef] [PubMed]
- van Erp, E.A.; Feyaerts, D.; Duijst, M.; Mulder, H.L.; Wicht, O.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Respiratory Syncytial Virus Infects Primary Neonatal and Adult Natural Killer Cells and Affects Their Antiviral Effector Function. J. Infect. Dis. 2019, 219, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Harker, J.A.; Godlee, A.; Wahlsten, J.L.; Lee, D.C.; Thorne, L.G.; Sawant, D.; Tregoning, J.S.; Caspi, R.R.; Bukreyev, A.; Collins, P.L.; et al. Interleukin 18 coexpression during respiratory syncytial virus infection results in enhanced disease mediated by natural killer cells. J. Virol. 2010, 84, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Hettinger, J.; Richards, D.M.; Hansson, J.; Barra, M.M.; Joschko, A.C.; Krijgsveld, J.; Feuerer, M. Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 2013, 14, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L. Monocyte subsets in man and other species. Cell Immunol. 2014, 289, 135–139. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Naidoo, K.K.; Ndumnego, O.C.; Ismail, N.; Dong, K.L.; Ndung’u, T. Antigen Presenting Cells Contribute to Persistent Immune Activation Despite Antiretroviral Therapy Initiation During Hyperacute HIV-1 Infection. Front. Immunol. 2021, 12, 738743. [Google Scholar] [CrossRef]
- Cao, W.; Taylor, A.K.; Biber, R.E.; Davis, W.G.; Kim, J.H.; Reber, A.J.; Chirkova, T.; De La Cruz, J.A.; Pandey, A.; Ranjan, P.; et al. Rapid differentiation of monocytes into type I IFN-producing myeloid dendritic cells as an antiviral strategy against influenza virus infection. J. Immunol. 2012, 189, 2257–2265. [Google Scholar] [CrossRef]
- Gill, M.A.; Long, K.; Kwon, T.; Muniz, L.; Mejias, A.; Connolly, J.; Roy, L.; Banchereau, J.; Ramilo, O. Differential recruitment of dendritic cells and monocytes to respiratory mucosal sites in children with influenza virus or respiratory syncytial virus infection. J. Infect. Dis. 2008, 198, 1667–1676. [Google Scholar] [CrossRef]
- Goritzka, M.; Makris, S.; Kausar, F.; Durant, L.R.; Pereira, C.; Kumagai, Y.; Culley, F.J.; Mack, M.; Akira, S.; Johansson, C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015, 212, 699–714. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, C.W.; Oh, D.S.; Jung, H.E.; Lee, H.K. Monocytes Contribute to IFN-beta Production via the MyD88-Dependent Pathway and Cytotoxic T-Cell Responses against Mucosal Respiratory Syncytial Virus Infection. Immune Netw. 2021, 21, e27. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.E.; Pedigo, H.; Johnson, T.R.; Shepherd, V.L. Surfactant protein-A enhances uptake of respiratory syncytial virus by monocytes and U937 macrophages. Am. J. Respir. Cell Mol. Biol. 2000, 23, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Ahout, I.M.; Jans, J.; Haroutiounian, L.; Simonetti, E.R.; van der Gaast-de Jongh, C.; Diavatopoulos, D.A.; de Jonge, M.I.; de Groot, R.; Ferwerda, G. Reduced Expression of HLA-DR on Monocytes During Severe Respiratory Syncytial Virus Infections. Pediatr. Infect. Dis. J. 2016, 35, e89–e96. [Google Scholar] [CrossRef]
- Besteman, S.B.; Phung, E.; Raeven, H.H.M.; Amatngalim, G.D.; Rumpret, M.; Crabtree, J.; Schepp, R.M.; Rodenburg, L.W.; Siemonsma, S.G.; Verleur, N.; et al. Recurrent Respiratory Syncytial Virus Infection in a CD14-Deficient Patient. J. Infect. Dis. 2022, 226, 258–269. [Google Scholar] [CrossRef]
- Bont, L.; Heijnen, C.J.; Kavelaars, A.; van Aalderen, W.M.; Brus, F.; Draaisma, J.T.; Geelen, S.M.; Kimpen, J.L. Monocyte IL-10 production during respiratory syncytial virus bronchiolitis is associated with recurrent wheezing in a one-year follow-up study. Am. J. Respir. Crit. Care Med. 2000, 161, 1518–1523. [Google Scholar] [CrossRef]
- Midulla, F.; Huang, Y.T.; Gilbert, I.A.; Cirino, N.M.; McFadden, E.R., Jr.; Panuska, J.R. Respiratory syncytial virus infection of human cord and adult blood monocytes and alveolar macrophages. Am. Rev. Respir. Dis. 1989, 140, 771–777. [Google Scholar] [CrossRef]
- Krilov, L.R.; Hendry, R.M.; Godfrey, E.; McIntosh, K. Respiratory virus infection of peripheral blood monocytes: Correlation with ageing of cells and interferon production in vitro. J. Gen. Virol. 1987, 68, 1749–1753. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tsutsumi, H.; Osaki, M.; Sone, S.; Imai, S.; Chiba, S. Respiratory syncytial virus infection of neonatal monocytes stimulates synthesis of interferon regulatory factor 1 and interleukin-1beta (IL-1beta)-converting enzyme and secretion of IL-1beta. J. Virol. 1998, 72, 837–840. [Google Scholar] [CrossRef]
- Polack, F.P.; Irusta, P.M.; Hoffman, S.J.; Schiatti, M.P.; Melendi, G.A.; Delgado, M.F.; Laham, F.R.; Thumar, B.; Hendry, R.M.; Melero, J.A.; et al. The cysteine-rich region of respiratory syncytial virus attachment protein inhibits innate immunity elicited by the virus and endotoxin. Proc. Natl. Acad. Sci. USA 2005, 102, 8996–9001. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Xiao, K.; Tang, L.; Xie, L. Diversity of Macrophages in Lung Homeostasis and Diseases. Front. Immunol. 2021, 12, 753940. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, J.; Wang, X.; Yang, P.; Zhao, D. Alveolar macrophages and airway hyperresponsiveness associated with respiratory syncytial virus infection. Front. Immunol. 2022, 13, 1012048. [Google Scholar] [CrossRef]
- Makris, S.; Bajorek, M.; Culley, F.J.; Goritzka, M.; Johansson, C. Alveolar Macrophages Can Control Respiratory Syncytial Virus Infection in the Absence of Type I Interferons. J. Innate Immun. 2016, 8, 452–463. [Google Scholar] [CrossRef]
- Oh, D.S.; Kim, T.H.; Lee, H.K. Differential Role of Anti-Viral Sensing Pathway for the Production of Type I Interferon beta in Dendritic Cells and Macrophages Against Respiratory Syncytial Virus A2 Strain Infection. Viruses 2019, 11, 62. [Google Scholar] [CrossRef]
- Kolli, D.; Gupta, M.R.; Sbrana, E.; Velayutham, T.S.; Chao, H.; Casola, A.; Garofalo, R.P. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 2014, 51, 502–515. [Google Scholar] [CrossRef]
- Reed, J.L.; Brewah, Y.A.; Delaney, T.; Welliver, T.; Burwell, T.; Benjamin, E.; Kuta, E.; Kozhich, A.; McKinney, L.; Suzich, J.; et al. Macrophage impairment underlies airway occlusion in primary respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2008, 198, 1783–1793. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, B.; Cheon, I.S.; Goplen, N.P.; Jiang, L.; Zhang, R.; Peebles, R.S.; Mack, M.; Kaplan, M.H.; Limper, A.H.; et al. PPAR-gamma in Macrophages Limits Pulmonary Inflammation and Promotes Host Recovery following Respiratory Viral Infection. J. Virol. 2019, 93, e00030-19. [Google Scholar] [CrossRef]
- Shirey, K.A.; Pletneva, L.M.; Puche, A.C.; Keegan, A.D.; Prince, G.A.; Blanco, J.C.; Vogel, S.N. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010, 3, 291–300. [Google Scholar] [CrossRef]
- Franke-Ullmann, G.; Pförtner, C.; Walter, P.; Steinmüller, C.; Lohmann-Matthes, M.L.; Kobzik, L.; Freihorst, J. Alteration of pulmonary macrophage function by respiratory syncytial virus infection in vitro. J. Immunol. 1995, 154, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Panuska, J.R.; Cirino, N.M.; Midulla, F.; Despot, J.E.; McFadden, E.R., Jr.; Huang, Y.T. Productive infection of isolated human alveolar macrophages by respiratory syncytial virus. J. Clin. Investig. 1990, 86, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.D.; Antunes, K.H.; Muraro, S.P.; de Souza, G.F.; da Silva, A.G.; Felipe, J.S.; Zanetti, L.C.; Czepielewski, R.S.; Magnus, K.; Scotta, M.; et al. TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during respiratory syncytial virus infection. Eur. Respir. J. 2021, 57, 2003764. [Google Scholar] [CrossRef] [PubMed]
- Haeberle, H.A.; Takizawa, R.; Casola, A.; Brasie, A.R.; Dieterich, H.-J.; van Rooijen, N.; Gatalica, Z.; Garofalo, R.P. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J. Infect. Dis. 2002, 186, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Werling, D. Binding and entry of respiratory syncytial virus into host cells and initiation of the innate immune response. Cell Microbiol. 2003, 5, 671–680. [Google Scholar] [CrossRef]
- Becker, S.; Quay, J.; Soukup, J. Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. J. Immunol. 1991, 147, 4307–4312. [Google Scholar] [CrossRef]
- Tsutsumi, H.; Matsuda, K.; Sone, S.; Takeuchi, R.; Chiba, S. Respiratory syncytial virus-induced cytokine production by neonatal macrophages. Clin. Exp. Immunol. 1996, 106, 442–446. [Google Scholar] [CrossRef]
- Eichinger, K.M.; Egana, L.; Orend, J.G.; Resetar, E.; Anderson, K.B.; Patel, R.; Empey, K.M. Alveolar macrophages support interferon gamma-mediated viral clearance in RSV-infected neonatal mice. Respir. Res. 2015, 16, 122. [Google Scholar] [CrossRef]
- Stockwin, L.H.; McGonagle, D.; Martin, I.G.; Blair, G.E. Dendritic cells: Immunological sentinels with a central role in health and disease. Immunol. Cell Biol. 2000, 78, 91–102. [Google Scholar] [CrossRef]
- Zaslona, Z.; Wilhelm, J.; Cakarova, L.; Marsh, L.M.; Seeger, W.; Lohmeyer, J.; von Wulffen, W. Transcriptome profiling of primary murine monocytes, lung macrophages and lung dendritic cells reveals a distinct expression of genes involved in cell trafficking. Respir. Res. 2009, 10, 2. [Google Scholar] [CrossRef]
- Boltjes, A.; van Wijk, F. Human dendritic cell functional specialization in steady-state and inflammation. Front. Immunol. 2014, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Oh, D.S.; Jung, H.E.; Chang, J.; Lee, H.K. Plasmacytoid Dendritic Cells Contribute to the Production of IFN-beta via TLR7-MyD88-Dependent Pathway and CTL Priming during Respiratory Syncytial Virus Infection. Viruses 2019, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Klouwenberg, P.K.; Tan, L.; Werkman, W.; van Bleek, G.M.; Coenjaerts, F. The role of Toll-like receptors in regulating the immune response against respiratory syncytial virus. Crit. Rev. Immunol. 2009, 29, 531–550. [Google Scholar] [CrossRef] [PubMed]
- Lukens, M.V.; Kruijsen, D.; Coenjaerts, F.E.; Kimpen, J.L.; van Bleek, G.M. Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J. Virol. 2009, 83, 7235–7243. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.R.; Johnson, C.N.; Corbett, K.S.; Edwards, G.C.; Graham, B.S. Primary human mDC1, mDC2, and pDC dendritic cells are differentially infected and activated by respiratory syncytial virus. PLoS ONE 2011, 6, e16458. [Google Scholar] [CrossRef] [PubMed]
- Rudd, B.D.; Schaller, M.A.; Smit, J.J.; Kunkel, S.L.; Neupane, R.; Kelley, L.; Berlin, A.A.; Lukacs, N.W. MyD88-mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J. Immunol. 2007, 178, 5820–5827. [Google Scholar] [CrossRef]
- Schijf, M.A.; Lukens, M.V.; Kruijsen, D.; van Uden, N.O.; Garssen, J.; Coenjaerts, F.E.; Van’t Land, B.; van Bleek, G.M. Respiratory syncytial virus induced type I IFN production by pDC is regulated by RSV-infected airway epithelial cells, RSV-exposed monocytes and virus specific antibodies. PLoS ONE 2013, 8, e81695. [Google Scholar] [CrossRef]
- Bartz, H.; Türkel, O.; Hoffjan, S.; Rothoeft, T.; Gonschorek, A.; Schauer, U. Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-gamma in naïve T cells. Immunology 2003, 109, 49–57. [Google Scholar] [CrossRef]
- de Graaff, P.M.A.; de Jong, E.C.; van Capel, T.M.; van Dijk, M.E.A.; Roholl, P.J.M.; Boes, J.; Luytjes, W.; Kimpen, J.L.L.; van Bleek, G.M. Respiratory syncytial virus infection of monocyte-derived dendritic cells decreases their capacity to activate CD4 T cells. J. Immunol. 2005, 175, 5904–5911. [Google Scholar] [CrossRef]
- Cespedes, P.F.; Bueno, S.M.; Ramirez, B.A.; Gomez, R.S.; Riquelme, S.A.; Palavecino, C.E.; Mackern-Oberti, J.P.; Mora, J.E.; Depoil, D.; Sacristan, C.; et al. Surface expression of the hRSV nucleoprotein impairs immunological synapse formation with T cells. Proc. Natl. Acad. Sci. USA 2014, 111, E3214–E3223. [Google Scholar] [CrossRef] [PubMed]
- Lau-Kilby, A.W.; Turfkruyer, M.; Kehl, M.; Yang, L.; Buchholz, U.J.; Hickey, K.; Malloy, A.M.W. Type I IFN ineffectively activates neonatal dendritic cells limiting respiratory antiviral T-cell responses. Mucosal Immunol. 2020, 13, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Malloy, A.M.; Ruckwardt, T.J.; Morabito, K.M.; Lau-Kilby, A.W.; Graham, B.S. Pulmonary Dendritic Cell Subsets Shape the Respiratory Syncytial Virus-Specific CD8+ T Cell Immunodominance Hierarchy in Neonates. J. Immunol. 2017, 198, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; You, D.; Saravia, J.; Siefker, D.T.; Jaligama, S.; Lee, G.I.; Sallam, A.A.; Harding, J.N.; Cormier, S.A. IL-4Ralpha on dendritic cells in neonates and Th2 immunopathology in respiratory syncytial virus infection. J. Leukoc. Biol. 2017, 102, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.R.; McLellan, J.S.; Graham, B.S. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. J. Virol. 2012, 86, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Stambas, J.; Lu, C.; Tripp, R.A. Innate and adaptive immune responses in respiratory virus infection: Implications for the clinic. Expert. Rev. Respir. Med. 2020, 14, 1141–1147. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef]
- Boehm, T.; Swann, J.B. Origin and evolution of adaptive immunity. Annu. Rev. Anim. Biosci. 2014, 2, 259–283. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.Y. B Cell Development and Maturation. Adv. Exp. Med. Biol. 2020, 1254, 1–22. [Google Scholar] [CrossRef]
- Lam, J.H.; Smith, F.L.; Baumgarth, N. B Cell Activation and Response Regulation During Viral Infections. Viral Immunol. 2020, 33, 294–306. [Google Scholar] [CrossRef]
- Roman, M.; Calhoun, W.J.; Hinton, K.L.; Avendano, L.F.; Simon, V.; Escobar, A.M.; Gaggero, A.; Diaz, P.V. Respiratory syncytial virus infection in infants is associated with predominant Th-2-like response. Am. J. Respir. Crit. Care Med. 1997, 156, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Andreano, E.; Paciello, I.; Bardelli, M.; Tavarini, S.; Sammicheli, C.; Frigimelica, E.; Guidotti, S.; Torricelli, G.; Biancucci, M.; D’Oro, U.; et al. The respiratory syncytial virus (RSV) prefusion F-protein functional antibody repertoire in adult healthy donors. EMBO Mol. Med. 2021, 13, e14035. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.E.; Falsey, A.R.; Halliley, J.L.; Sanz, I.; Walsh, E.E. Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J. Infect. Dis. 2010, 202, 1659–1666. [Google Scholar] [CrossRef]
- Raes, M.; Alliet, P.; Gillis, P.; Kortleven, J.; Magerman, K.; Rummens, J.L. Peripheral blood T and B lymphocyte subpopulations in infants with acute respiratory syncytial virus brochiolitis. Pediatr. Allergy Immunol. 1997, 8, 97–102. [Google Scholar] [CrossRef]
- Xiao, X.; Tang, A.; Cox, K.S.; Wen, Z.; Callahan, C.; Sullivan, N.L.; Nahas, D.D.; Cosmi, S.; Galli, J.D.; Minnier, M.; et al. Characterization of potent RSV neutralizing antibodies isolated from human memory B cells and identification of diverse RSV/hMPV cross-neutralizing epitopes. MAbs 2019, 11, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Alturaiki, W.; McFarlane, A.J.; Rose, K.; Corkhill, R.; McNamara, P.S.; Schwarze, J.; Flanagan, B.F. Expression of the B cell differentiation factor BAFF and chemokine CXCL13 in a murine model of Respiratory Syncytial Virus infection. Cytokine 2018, 110, 267–271. [Google Scholar] [CrossRef]
- Reed, J.L.; Welliver, T.P.; Sims, G.P.; McKinney, L.; Velozo, L.; Avendano, L.; Hintz, K.; Luma, J.; Coyle, A.J.; Welliver, R.C., Sr. Innate Immune Signals Modulate Antiviral and Polyreactive Antibody Responses during Severe Respiratory Syncytial Virus Infection. J. Infect. Dis. 2009, 199, 1128–1138. [Google Scholar] [CrossRef]
- Falsey, A.R.; Singh, H.K.; Walsh, E.E. Serum antibody decay in adults following natural respiratory syncytial virus infection. J. Med. Virol. 2006, 78, 1493–1497. [Google Scholar] [CrossRef]
- Green, C.A.; Sande, C.J.; de Lara, C.; Thompson, A.J.; Silva-Reyes, L.; Napolitano, F.; Pierantoni, A.; Capone, S.; Vitelli, A.; Klenerman, P.; et al. Humoral and cellular immunity to RSV in infants, children and adults. Vaccine 2018, 36, 6183–6190. [Google Scholar] [CrossRef]
- Habibi, M.S.; Jozwik, A.; Makris, S.; Dunning, J.; Paras, A.; DeVincenzo, J.P.; de Haan, C.A.; Wrammert, J.; Openshaw, P.J.; Chiu, C.; et al. Impaired Antibody-mediated Protection and Defective IgA B-Cell Memory in Experimental Infection of Adults with Respiratory Syncytial Virus. Am. J. Respir. Crit. Care Med. 2015, 191, 1040–1049. [Google Scholar] [CrossRef]
- Cortjens, B.; Yasuda, E.; Yu, X.; Wagner, K.; Claassen, Y.B.; Bakker, A.Q.; van Woensel, J.B.M.; Beaumont, T. Broadly Reactive Anti-Respiratory Syncytial Virus G Antibodies from Exposed Individuals Effectively Inhibit Infection of Primary Airway Epithelial Cells. J. Virol. 2017, 91, e02357-16. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.E.; Meyerholz, D.K.; Varga, S.M. Pre-existing neutralizing antibodies prevent CD8 T cell-mediated immunopathology following respiratory syncytial virus infection. Mucosal Immunol. 2020, 13, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E. Relationship of serum antibody to risk of respiratory syncytial virus infection in elderly adults. J. Infect. Dis. 1998, 177, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E.; Falsey, A.R. Humoral and mucosal immunity in protection from natural respiratory syncytial virus infection in adults. J. Infect. Dis. 2004, 190, 373–378. [Google Scholar] [CrossRef]
- Walsh, E.E.; Peterson, D.R.; Falsey, A.R. Risk factors for severe respiratory syncytial virus infection in elderly persons. J. Infect. Dis. 2004, 189, 233–238. [Google Scholar] [CrossRef]
- Ascough, S.; Dayananda, P.; Kalyan, M.; Kuong, S.U.; Gardener, Z.; Bergstrom, E.; Paterson, S.; Kar, S.; Avadhan, V.; Thwaites, R.; et al. Divergent age-related humoral correlates of protection against respiratory syncytial virus infection in older and young adults: A pilot, controlled, human infection challenge model. Lancet Healthy Longev. 2022, 3, e405–e416. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E. Humoral immunity to respiratory syncytial virus infection in the elderly. J. Med. Virol. 1992, 36, 39–43. [Google Scholar] [CrossRef]
- Walsh, E.E.; Peterson, D.R.; Kalkanoglu, A.E.; Lee, F.E.; Falsey, A.R. Viral shedding and immune responses to respiratory syncytial virus infection in older adults. J. Infect. Dis. 2013, 207, 1424–1432. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E.; Looney, R.J.; Kolassa, J.E.; Formica, M.A.; Criddle, M.C.; Hall, W.J. Comparison of respiratory syncytial virus humoral immunity and response to infection in young and elderly adults. J. Med. Virol. 1999, 59, 221–226. [Google Scholar] [CrossRef]
- Walsh, E.E.; Falsey, A.R. Age related differences in humoral immune response to respiratory syncytial virus infection in adults. J. Med. Virol. 2004, 73, 295–299. [Google Scholar] [CrossRef]
- Goodwin, E.; Gilman, M.S.A.; Wrapp, D.; Chen, M.; Ngwuta, J.O.; Moin, S.M.; Bai, P.; Sivasubramanian, A.; Connor, R.I.; Wright, P.F.; et al. Infants Infected with Respiratory Syncytial Virus Generate Potent Neutralizing Antibodies that Lack Somatic Hypermutation. Immunity 2018, 48, 339–349 e335. [Google Scholar] [CrossRef]
- van Erp, E.A.; Lakerveld, A.J.; de Graaf, E.; Larsen, M.D.; Schepp, R.M.; Hipgrave Ederveen, A.L.; Ahout, I.M.; de Haan, C.A.; Wuhrer, M.; Luytjes, W.; et al. Natural killer cell activation by respiratory syncytial virus-specific antibodies is decreased in infants with severe respiratory infections and correlates with Fc-glycosylation. Clin. Transl. Immunol. 2020, 9, e1112. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.V.; Weitkamp, J.H.; Blum, D.L.; LaFleur, B.J.; Crowe, J.E., Jr. The human neonatal B cell response to respiratory syncytial virus uses a biased antibody variable gene repertoire that lacks somatic mutations. Mol. Immunol. 2009, 47, 407–414. [Google Scholar] [CrossRef]
- Murphy, B.R.; Graham, B.S.; Prince, G.A.; Walsh, E.E.; Chanock, R.M.; Karzon, D.T.; Wright, P.F. Serum and nasal-wash immunoglobulin G and A antibody response of infants and children to respiratory syncytial virus F and G glycoproteins following primary infection. J. Clin. Microbiol. 1986, 23, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Kruijsen, D.; Bakkers, M.J.; van Uden, N.O.; Viveen, M.C.; van der Sluis, T.C.; Kimpen, J.L.; Leusen, J.H.; Coenjaerts, F.E.; van Bleek, G.M. Serum antibodies critically affect virus-specific CD4+/CD8+ T cell balance during respiratory syncytial virus infections. J. Immunol. 2010, 185, 6489–6498. [Google Scholar] [CrossRef] [PubMed]
- Vono, M.; Eberhardt, C.S.; Auderset, F.; Mastelic-Gavillet, B.; Lemeille, S.; Christensen, D.; Andersen, P.; Lambert, P.H.; Siegrist, C.A. Maternal Antibodies Inhibit Neonatal and Infant Responses to Vaccination by Shaping the Early-Life B Cell Repertoire within Germinal Centers. Cell Rep. 2019, 28, 1773–1784 e1775. [Google Scholar] [CrossRef]
- Zhivaki, D.; Lemoine, S.; Lim, A.; Morva, A.; Vidalain, P.O.; Schandene, L.; Casartelli, N.; Rameix-Welti, M.A.; Herve, P.L.; Deriaud, E.; et al. Respiratory Syncytial Virus Infects Regulatory B Cells in Human Neonates via Chemokine Receptor CX3CR1 and Promotes Lung Disease Severity. Immunity 2017, 46, 301–314. [Google Scholar] [CrossRef]
- Bukreyev, A.; Yang, L.; Fricke, J.; Cheng, L.; Ward, J.M.; Murphy, B.R.; Collins, P.L. The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on Fc receptor-bearing leukocytes. J. Virol. 2008, 82, 12191–12204. [Google Scholar] [CrossRef]
- Groothuis, J.R.; Levin, M.J.; Rodriguez, W.; Hall, C.B.; Long, C.E.; Kim, H.W.; Lauer, B.A.; Hemming, V.G. Use of intravenous gamma globulin to passively immunize high-risk children against respiratory syncytial virus: Safety and pharmacokinetics. The RSVIG Study Group. Antimicrob. Agents Chemother. 1991, 35, 1469–1473. [Google Scholar] [CrossRef]
- Groothuis, J.R.; Simoes, E.A.; Hemming, V.G. Respiratory syncytial virus (RSV) infection in preterm infants and the protective effects of RSV immune globulin (RSVIG). Respiratory Syncytial Virus Immune Globulin Study Group. Pediatrics 1995, 95, 463–467. [Google Scholar]
- Rodriguez, W.J.; Gruber, W.C.; Groothuis, J.R.; Simoes, E.A.; Rosas, A.J.; Lepow, M.; Kramer, A.; Hemming, V. Respiratory syncytial virus immune globulin treatment of RSV lower respiratory tract infection in previously healthy children. Pediatrics 1997, 100, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Nirsevimab: First Approval. Drugs 2023, 83, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Group, I.-R.S. Palivizumab, a Humanized Respiratory Syncytial Virus Monoclonal Antibody, Reduces Hospitalization From Respiratory Syncytial Virus Infection in High-risk Infants. Pediatrics 1998, 102, 531–537. [Google Scholar] [CrossRef]
- Kalia, V.; Sarkar, S.; Gourley, T.S.; Rouse, B.T.; Ahmed, R. Differentiation of memory B and T cells. Curr. Opin. Immunol. 2006, 18, 255–264. [Google Scholar] [CrossRef]
- Palacios-Pedrero, M.A.; Jansen, J.M.; Blume, C.; Stanislawski, N.; Jonczyk, R.; Molle, A.; Hernandez, M.G.; Kaiser, F.K.; Jung, K.; Osterhaus, A.; et al. Signs of immunosenescence correlate with poor outcome of mRNA COVID-19 vaccination in older adults. Nat. Aging 2022, 2, 896–905. [Google Scholar] [CrossRef]
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef]
- Benova, K.; Hanckova, M.; Koci, K.; Kudelova, M.; Betakova, T. T cells and their function in the immune response to viruses. Acta Virol. 2020, 64, 131–143. [Google Scholar] [CrossRef]
- Sealy, R.E.; Surman, S.L.; Hurwitz, J.L. CD4(+) T cells support establishment of RSV-specific IgG and IgA antibody secreting cells in the upper and lower murine respiratory tract following RSV infection. Vaccine 2017, 35, 2617–2621. [Google Scholar] [CrossRef]
- Kinnear, E.; Lambert, L.; McDonald, J.U.; Cheeseman, H.M.; Caproni, L.J.; Tregoning, J.S. Airway T cells protect against RSV infection in the absence of antibody. Mucosal Immunol. 2018, 11, 249–256. [Google Scholar] [CrossRef]
- Guvenel, A.; Jozwik, A.; Ascough, S.; Ung, S.K.; Paterson, S.; Kalyan, M.; Gardener, Z.; Bergstrom, E.; Kar, S.; Habibi, M.S.; et al. Epitope-specific airway-resident CD4+ T cell dynamics during experimental human RSV infection. J. Clin. Investig. 2020, 130, 523–538. [Google Scholar] [CrossRef]
- Kitcharoensakkul, M.; Bacharier, L.B.; Yin-Declue, H.; Boomer, J.S.; Sajol, G.; Leung, M.K.; Wilson, B.; Schechtman, K.B.; Atkinson, J.P.; Green, J.M.; et al. Impaired tumor necrosis factor-alpha secretion by CD4 T cells during respiratory syncytial virus bronchiolitis associated with recurrent wheeze. Immun. Inflamm. Dis. 2020, 8, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Welliver, T.P.; Reed, J.L.; Welliver, R.C., Sr. Respiratory syncytial virus and influenza virus infections: Observations from tissues of fatal infant cases. Pediatr. Infect. Dis. J. 2008, 27, S92–S96. [Google Scholar] [CrossRef] [PubMed]
- De Swart, R.L.; Kuiken, T.; Timmerman, H.H.; van Amerongen, G.; Van Den Hoogen, B.G.; Vos, H.W.; Neijens, H.J.; Andeweg, A.C.; Osterhaus, A.D. Immunization of macaques with formalin-inactivated respiratory syncytial virus (RSV) induces interleukin-13-associated hypersensitivity to subsequent RSV infection. J. Virol. 2002, 76, 11561–11569. [Google Scholar] [CrossRef]
- Waris, M.E.; Tsou, C.; Erdman, D.D.; Zaki, S.R.; Anderson, L.J. Respiratory synctial virus infection in BALB/c mice previously immunized with formalin-inactivated virus induces enhanced pulmonary inflammatory response with a predominant Th2-like cytokine pattern. J. Virol. 1996, 70, 2852–2860. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.J.; Hartwig, S.M.; Meyerholz, D.K.; Varga, S.M. RSV vaccine-enhanced disease is orchestrated by the combined actions of distinct CD4 T cell subsets. PLoS Pathog. 2015, 11, e1004757. [Google Scholar] [CrossRef]
- You, D.; Marr, N.; Saravia, J.; Shrestha, B.; Lee, G.I.; Turvey, S.E.; Brombacher, F.; Herbert, D.R.; Cormier, S.A. IL-4Ralpha on CD4+ T cells plays a pathogenic role in respiratory syncytial virus reinfection in mice infected initially as neonates. J. Leukoc. Biol. 2013, 93, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Roumanes, D.; Falsey, A.R.; Quataert, S.; Secor-Socha, S.; Lee, F.E.; Yang, H.; Bandyopadhyay, S.; Holden-Wiltse, J.; Topham, D.J.; Walsh, E.E. T-Cell Responses in Adults During Natural Respiratory Syncytial Virus Infection. J. Infect. Dis. 2018, 218, 418–428. [Google Scholar] [CrossRef]
- Graham, B.S.; Bunton, L.A.; Wright, P.F.; Karzon, D.T. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Investig. 1991, 88, 1026–1033. [Google Scholar] [CrossRef]
- Conners, M.; Kulkarni, A.B.; Firestone, C.Y.; Holmes, K.L.; Morse, H.C.r.; Sotnikov, A.V.; Murphy, B.R. Pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of formalin-inactivated RSV-immunized BALB/c mice is abrogated by depletion of CD4+ T cells. J. Virol. 1992, 66, 7444–7451. [Google Scholar] [CrossRef]
- Schneider-Ohrum, K.; Snell Bennett, A.; Rajani, G.M.; Hostetler, L.; Maynard, S.K.; Lazzaro, M.; Cheng, L.I.; O’Day, T.; Cayatte, C. CD4(+) T Cells Drive Lung Disease Enhancement Induced by Immunization with Suboptimal Doses of Respiratory Syncytial Virus Fusion Protein in the Mouse Model. J. Virol. 2019, 93, e00695-19. [Google Scholar] [CrossRef]
- Raiden, S.; Sananez, I.; Remes-Lenicov, F.; Pandolfi, J.; Romero, C.; De Lillo, L.; Ceballos, A.; Geffner, J.; Arruvito, L. Respiratory Syncytial Virus (RSV) Infects CD4+ T Cells: Frequency of Circulating CD4+ RSV+ T Cells as a Marker of Disease Severity in Young Children. J. Infect. Dis. 2017, 215, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Schlender, J.; Walliser, G.; Fricke, J.; Conzelmann, K.K. Respiratory syncytial virus fusion protein mediates inhibition of mitogen-induced T-cell proliferation by contact. J. Virol. 2002, 76, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Bont, L. Natural Reinfection with Respiratory Syncytial Virus Does Not Boost Virus-Specific T-Cell Immunity. Pediatr. Res. 2002, 52, 363–367. [Google Scholar] [CrossRef]
- Heidema, J.; Lukens, M.V.; van Maren, W.W.C.; van Dijk, M.E.A.; Otten, H.G.; van Vught, A.J.; van der Werff, D.B.M.; van Gestel, S.J.P.; Semple, M.G.; Smyth, R.L.; et al. CD8+ T cell responses in bronchoalveolar lavage fluid and peripheral blood mononuclear cells of infants with severe primary respiratory syncytial virus infections. J. Immunol. 2007, 179, 8410–8417. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.J.; Weiss, K.A.; Hartwig, S.M.; Varga, S.M. The pulmonary localization of virus-specific T lymphocytes is governed by the tissue tropism of infection. J. Virol. 2014, 88, 9010–9016. [Google Scholar] [CrossRef]
- Jozwik, A.; Habibi, M.S.; Paras, A.; Zhu, J.; Guvenel, A.; Dhariwal, J.; Almond, M.; Wong, E.H.C.; Sykes, A.; Maybeno, M.; et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat. Commun. 2015, 6, 10224. [Google Scholar] [CrossRef]
- Ruckwardt, T.J.; Luongo, C.; Malloy, A.M.; Liu, J.; Chen, M.; Collins, P.L.; Graham, B.S. Responses against a subdominant CD8+ T cell epitope protect against immunopathology caused by a dominant epitope. J. Immunol. 2010, 185, 4673–4680. [Google Scholar] [CrossRef]
- Taylor, G.; Stott, E.J.; Hayle, A.J. Cytotoxic lymphocytes in the lungs of mice infected with respiratory syncytial virus. J. Gen. Virol. 1985, 66, 2533–2538. [Google Scholar] [CrossRef]
- Aberle, J.H.; Aberle, S.W.; Dworzak, M.N.; Mandl, C.W.; Rebhandl, W.; Vollnhofer, G.; Kundi, M.; Popow-Kraupp, T. Reduced interferon-gamma expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 1999, 160, 1263–1268. [Google Scholar] [CrossRef]
- De Weerd, W.; Twilhaar, W.N.; Kimpen, J.L. T cell subset analysis in peripheral blood of children with RSV bronchiolitis. Scand. J. Infect. Dis. 1998, 30, 77–80. [Google Scholar] [CrossRef]
- El Saleeby, C.M.; Suzich, J.; Conley, M.E.; DeVincenzo, J.P. Quantitative effects of palivizumab and donor-derived T cells on chronic respiratory syncytial virus infection, lung disease, and fusion glycoprotein amino acid sequences in a patient before and after bone marrow transplantation. Clin. Infect. Dis. 2004, 39, 17–20. [Google Scholar] [CrossRef]
- Heidema, J.; de Bree, G.J.; de Graaff, P.M.A.; van Maren, W.W.C.; Hoogerhout, P.; Out, T.A.; Kimpen, J.L.L.; van Bleek, G.M. Human CD8(+) T cell responses against five newly identified respiratory syncytial virus-derived epitopes. J. Gen. Virol. 2004, 85, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Lukens, M.V.; Claassen, E.A.; de Graaff, P.M.; van Dijk, M.E.; Hoogerhout, P.; Toebes, M.; Schumacher, T.N.; van der Most, R.G.; Kimpen, J.L.; van Bleek, G.M. Characterization of the CD8+ T cell responses directed against respiratory syncytial virus during primary and secondary infection in C57BL/6 mice. Virology 2006, 352, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Ostler, T.; Ehl, S. Pulmonary T cells induced by respiratory syncytial virus are functional and can make an important contribution to long-lived protective immunity. Eur. J. Immunol. 2002, 32, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Ruckwardt, T.J.; Bonaparte, K.L.; Nason, M.C.; Graham, B.S. Regulatory T cells promote early influx of CD8+ T cells in the lungs of respiratory syncytial virus-infected mice and diminish immunodominance disparities. J. Virol. 2009, 83, 3019–3028. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Braciale, T.J. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 1997, 186, 421–432. [Google Scholar] [CrossRef]
- Ostler, T.; Davidson, W.; Ehl, S. Virus clearance and immunopathology by CD8(+) T cells during infection with respiratory syncytial virus are mediated by IFN-gamma. Eur. J. Immunol. 2002, 32, 2117–2123. [Google Scholar] [CrossRef]
- Bem, R.A.; Bos, A.P.; Bots, M.; Wolbink, A.M.; van Ham, S.M.; Medema, J.P.; Lutter, R.; van Woensel, J.B.M. Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr. Res. 2008, 63, 650–655. [Google Scholar] [CrossRef]
- Siefker, D.T.; Vu, L.; You, D.; McBride, A.; Taylor, R.; Jones, T.L.; DeVincenzo, J.; Cormier, S.A. Respiratory Syncytial Virus Disease Severity Is Associated with Distinct CD8(+) T-Cell Profiles. Am. J. Respir. Crit. Care Med. 2020, 201, 325–334. [Google Scholar] [CrossRef]
- van Schaik, S.M.; Obot, N.; Enhorning, G.; Hintz, K.; Gross, K.; Hancock, G.E.; Stack, A.M.; Welliver, R.C. Role of interferon gamma in the pathogenesis of primary respiratory syncytial virus infection in BALB/c mice. J. Med. Virol. 2000, 62, 257–266. [Google Scholar] [CrossRef]
- Rutigliano, J.A.; Graham, B.S. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J. Immunol. 2004, 173, 3408–3417. [Google Scholar] [CrossRef] [PubMed]
- Meckiff, B.J.; Ramirez-Suastegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell 2020, 183, 1340–1353 e1316. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.T.; Wang, Z.L.; Tian, P.; Gong, X.N.; Fan, Y.C.; Wang, K. Treg/Th17 imbalance and its clinical significance in patients with hepatitis B-associated liver cirrhosis. Diagn. Pathol. 2019, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.A. Role of regulatory T cells in human diseases. J. Allergy Clin. Immunol. 2005, 116, 949–959, quiz 960. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Menard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Lewkowicz, P.; Lewkowicz, N.; Sasiak, A.; Tchorzewski, H. Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J. Immunol. 2006, 177, 7155–7163. [Google Scholar] [CrossRef]
- Rudensky, A.V. Regulatory T cells and Foxp3. Immunol. Rev. 2011, 241, 260–268. [Google Scholar] [CrossRef]
- Weiss, K.A.; Christiaansen, A.F.; Fulton, R.B.; Meyerholz, D.K.; Varga, S.M. Multiple CD4+ T cell subsets produce immunomodulatory IL-10 during respiratory syncytial virus infection. J. Immunol. 2011, 187, 3145–3154. [Google Scholar] [CrossRef]
- Demoulins, T.; Brugger, M.; Zumkehr, B.; Oliveira Esteves, B.I.; Mehinagic, K.; Fahmi, A.; Borcard, L.; Taddeo, A.; Jandrasits, D.; Posthaus, H.; et al. The specific features of the developing T cell compartment of the neonatal lung are a determinant of respiratory syncytial virus immunopathogenesis. PLoS Pathog. 2021, 17, e1009529. [Google Scholar] [CrossRef]
- Fulton, R.B.; Meyerholz, D.K.; Varga, S.M. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J. Immunol. 2010, 185, 2382–2392. [Google Scholar] [CrossRef]
- Lee, D.C.; Harker, J.A.; Tregoning, J.S.; Atabani, S.F.; Johansson, C.; Schwarze, J.; Openshaw, P.J. CD25+ natural regulatory T cells are critical in limiting innate and adaptive immunity and resolving disease following respiratory syncytial virus infection. J. Virol. 2010, 84, 8790–8798. [Google Scholar] [CrossRef] [PubMed]
- Loebbermann, J.; Thornton, H.; Durant, L.; Sparwasser, T.; Webster, K.E.; Sprent, J.; Culley, F.J.; Johansson, C.; Openshaw, P.J. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. 2012, 5, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.Y.; Huang, J.Y.; Lin, Y.W.; Yu, S.L.; Chitra, E.; Chang, C.K.; Sung, W.C.; Chong, P.; Chow, Y.H. Depletion of regulatory T-cells leads to moderate B-cell antigenicity in respiratory syncytial virus infection. Int. J. Infect. Dis. 2015, 41, 56–64. [Google Scholar] [CrossRef]
- Loebbermann, J.; Schnoeller, C.; Thornton, H.; Durant, L.; Sweeney, N.P.; Schuijs, M.; O’Garra, A.; Johansson, C.; Openshaw, P.J. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS ONE 2012, 7, e32371. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cardani, A.; Sharma, A.K.; Laubach, V.E.; Jack, R.S.; Muller, W.; Braciale, T.J. Autocrine regulation of pulmonary inflammation by effector T-cell derived IL-10 during infection with respiratory syncytial virus. PLoS Pathog. 2011, 7, e1002173. [Google Scholar] [CrossRef]
- Christiaansen, A.F.; Syed, M.A.; Ten Eyck, P.P.; Hartwig, S.M.; Durairaj, L.; Kamath, S.S.; Varga, S.M. Altered Treg and cytokine responses in RSV-infected infants. Pediatr. Res. 2016, 80, 702–709. [Google Scholar] [CrossRef]
- Raiden, S.; Pandolfi, J.; Payaslian, F.; Anderson, M.; Rivarola, N.; Ferrero, F.; Urtasun, M.; Fainboim, L.; Geffner, J.; Arruvito, L. Depletion of circulating regulatory T cells during severe respiratory syncytial virus infection in young children. Am. J. Respir. Crit. Care Med. 2014, 189, 865–868. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Khare, A.; Oriss, T.B.; Raundhal, M.; Morse, C.; Yarlagadda, M.; Wenzel, S.E.; Moore, M.L.; Peebles, R.S., Jr.; Ray, A.; et al. Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat. Med. 2012, 18, 1525–1530. [Google Scholar] [CrossRef]
- Badovinac, V.P.; Porter, B.B.; Harty, J.T. CD8+ T cell contraction is controlled by early inflammation. Nat. Immunol. 2004, 5, 809–817. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Turner, D.L.; Bickham, K.L.; Thome, J.J.; Kim, C.Y.; D’Ovidio, F.; Wherry, E.J.; Farber, D.L. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014, 7, 501–510. [Google Scholar] [CrossRef]
- Zheng, M.Z.M.; Wakim, L.M. Tissue resident memory T cells in the respiratory tract. Mucosal Immunol. 2022, 15, 379–388. [Google Scholar] [CrossRef] [PubMed]
- De Bree, G.J.; Heidema, J.; van Leeuwen, E.M.M.; van Bleek, G.M.; Jonkers, R.E.; Jansen, H.M.; van Lier, R.A.W.; Out, T.A. Respiratory syncytial virus-specific CD8+ memory T cell responses in elderly persons. J. Infect. Dis. 2005, 191, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Varese, A.; Nakawesi, J.; Farias, A.; Kirsebom, F.C.M.; Paulsen, M.; Nuriev, R.; Johansson, C. Type I interferons and MAVS signaling are necessary for tissue resident memory CD8+ T cell responses to RSV infection. PLoS Pathog. 2022, 18, e1010272. [Google Scholar] [CrossRef] [PubMed]
- Heidema, J.; Rossen, J.W.A.; Lukens, M.V.; Ketel, M.S.; Scheltens, E.; Kranendonk, M.E.G.; van Maren, W.W.C.; van Loon, A.M.; Otten, H.G.; Kimpen, J.L.L.; et al. Dynamics of human respiratory virus-specific CD8+ T cell responses in blood and airways during episodes of common cold. J. Immunol. 2008, 181, 5551–5559. [Google Scholar] [CrossRef] [PubMed]
- Luangrath, M.A.; Schmidt, M.E.; Hartwig, S.M.; Varga, S.M. Tissue-Resident Memory T Cells in the Lungs Protect against Acute Respiratory Syncytial Virus Infection. Immunohorizons 2021, 5, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Braciale, T.J. Respiratory syncytial virus infection suppresses lung CD8+ Tcell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nat. Med. 2002, 8, 54–60. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Knudson, C.J.; Hartwig, S.M.; Pewe, L.L.; Meyerholz, D.K.; Langlois, R.A.; Harty, J.T.; Varga, S.M. Memory CD8 T cells mediate severe immunopathology following respiratory syncytial virus infection. PLoS Pathog. 2018, 14, e1006810. [Google Scholar] [CrossRef]
- Chirkova, T.; Rosas-Salazar, C.; Gebretsadik, T.; Jadhao, S.J.; Chappell, J.D.; Peebles, R.S., Jr.; Dupont, W.D.; Newcomb, D.C.; Berdnikovs, S.; Gergen, P.J.; et al. Effect of Infant RSV Infection on Memory T Cell Responses at Age 2-3 Years. Front. Immunol. 2022, 13, 826666. [Google Scholar] [CrossRef]
- Zens, K.D.; Chen, J.K.; Guyer, R.S.; Wu, F.L.; Cvetkovski, F.; Miron, M.; Farber, D.L. Reduced generation of lung tissue-resident memory T cells during infancy. J. Exp. Med. 2017, 214, 2915–2932. [Google Scholar] [CrossRef]
- Chanock, R.; Roizman, B.; Myers, R. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). I. Isolation, properties and characterization. Am. J. Hyg. 1957, 66, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Blount, R.E.J.; Morris, J.A.; Savage, R.E. Recovery of cytopathogenic agent from chimpanzees with coryza. Proc. Soc. Exp. Biol. Med. 1956, 92, 544–594. [Google Scholar] [CrossRef] [PubMed]
- Crank, M.C.; Ruckwardt, T.J.; Chen, M.; Morabito, K.M.; Phung, E.; Costner, P.J.; Holman, L.A.; Hickman, S.P.; Berkowitz, N.M.; Gordon, I.J.; et al. A proof of concept for structure-based vaccine design targeting RSV in humans. Science 2019, 365, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Okuda, K.; Shimada, M. Developments in Viral Vector-Based Vaccines. Vaccines 2014, 2, 624–641. [Google Scholar] [CrossRef]
- Jordan, E.; Lawrence, S.J.; Meyer, T.P.H.; Schmidt, D.; Schultz, S.; Mueller, J.; Stroukova, D.; Koenen, B.; Gruenert, R.; Silbernagl, G.; et al. Broad Antibody and Cellular Immune Response From a Phase 2 Clinical Trial With a Novel Multivalent Poxvirus-Based Respiratory Syncytial Virus Vaccine. J. Infect. Dis. 2021, 223, 1062–1072. [Google Scholar] [CrossRef]
- Samy, N.; Reichhardt, D.; Schmidt, D.; Chen, L.M.; Silbernagl, G.; Vidojkovic, S.; Meyer, T.P.; Jordan, E.; Adams, T.; Weidenthaler, H.; et al. Safety and immunogenicity of novel modified vaccinia Ankara-vectored RSV vaccine: A randomized phase I clinical trial. Vaccine 2020, 38, 2608–2619. [Google Scholar] [CrossRef]
- Falsey, A.R.; Williams, K.; Gymnopoulou, E.; Bart, S.; Ervin, J.; Bastian, A.R.; Menten, J.; De Paepe, E.; Vandenberghe, S.; Chan, E.K.H.; et al. Efficacy and Safety of an Ad26.RSV.preF-RSV preF Protein Vaccine in Older Adults. N. Engl. J. Med. 2023, 388, 609–620. [Google Scholar] [CrossRef]
- Stuart, A.S.V.; Virta, M.; Williams, K.; Seppa, I.; Hartvickson, R.; Greenland, M.; Omoruyi, E.; Bastian, A.R.; Haazen, W.; Salisch, N.; et al. Phase 1/2a Safety and Immunogenicity of an Adenovirus 26 Vector Respiratory Syncytial Virus (RSV) Vaccine Encoding Prefusion F in Adults 18-50 Years and RSV-Seropositive Children 12-24 Months. J. Infect. Dis. 2022, 227, 71–82. [Google Scholar] [CrossRef]
- Sadoff, J.; De Paepe, E.; DeVincenzo, J.; Gymnopoulou, E.; Menten, J.; Murray, B.; Rosemary Bastian, A.; Vandebosch, A.; Haazen, W.; Noulin, N.; et al. Prevention of Respiratory Syncytial Virus Infection in Healthy Adults by a Single Immunization of Ad26.RSV.preF in a Human Challenge Study. J. Infect. Dis. 2022, 226, 396–406. [Google Scholar] [CrossRef]
- Baxter, D. Active and passive immunity, vaccine types, excipients and licensing. Occup. Med. 2007, 57, 552–556. [Google Scholar] [CrossRef]
- Walsh, E.E.; Falsey, A.R.; Scott, D.A.; Gurtman, A.; Zareba, A.M.; Jansen, K.U.; Gruber, W.C.; Dormitzer, P.R.; Swanson, K.A.; Radley, D.; et al. A Randomized Phase 1/2 Study of a Respiratory Syncytial Virus Prefusion F Vaccine. J. Infect. Dis. 2022, 225, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E.; Scott, D.A.; Gurtman, A.; Zareba, A.; Jansen, K.U.; Gruber, W.C.; Dormitzer, P.R.; Swanson, K.A.; Jiang, Q.; et al. Phase 1/2 Randomized Study of the Immunogenicity, Safety, and Tolerability of a Respiratory Syncytial Virus Prefusion F Vaccine in Adults With Concomitant Inactivated Influenza Vaccine. J. Infect. Dis. 2022, 225, 2056–2066. [Google Scholar] [CrossRef] [PubMed]
- Schmoele-Thoma, B.; Zareba, A.M.; Jiang, Q.; Maddur, M.S.; Danaf, R.; Mann, A.; Eze, K.; Fok-Seang, J.; Kabir, G.; Catchpole, A.; et al. Vaccine Efficacy in Adults in a Respiratory Syncytial Virus Challenge Study. N. Engl. J. Med. 2022, 386, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E.; Pérez Marc, G.; Zareba, A.M.; Falsey, A.R.; Jiang, Q.; Patton, M.; Polack, F.P.; Llapur, C.; Doreski, P.A.; Ilangovan, K.; et al. Efficacy and Safety of a Bivalent RSV Prefusion F Vaccine in Older Adults. N. Engl. J. Med. 2023, 388, 1465–1477. [Google Scholar] [CrossRef]
- Kampmann, B.; Madhi, S.A.; Munjal, I.; Simões, E.A.F.; Pahud, B.A.; Llapur, C.; Baker, J.; Pérez Marc, G.; Radley, D.; Shittu, E.; et al. Bivalent Prefusion F Vaccine in Pregnancy to Prevent RSV Illness in Infants. N. Engl. J. Med. 2023, 388, 1451–1464. [Google Scholar] [CrossRef]
- Simoes, E.A.F. Respiratory Syncytial Virus Disease in Young Children and Older Adults in Europe: A Burden and Economic Perspective. J. Infect. Dis. 2022, 226, S1–S9. [Google Scholar] [CrossRef]
- Bebia, Z.; Reyes, O.; Jeanfreau, R.; Kantele, A.; De Leon, R.G.; Garcia Sanchez, M.; Banooni, P.; Gardener, G.J.; Bartha Rasero, J.L.; Encinas Pardilla, M.B.; et al. Safety and immunogenicity of an investigational respiratory syncytial virus vaccine (RSVPreF3) in mothers and their infants: A phase 2 randomized trial. J. Infect. Dis. 2023, 228, 299–310. [Google Scholar] [CrossRef]
- Papi, A.; Ison, M.G.; Langley, J.M.; Lee, D.G.; Leroux-Roels, I.; Martinon-Torres, F.; Schwarz, T.F.; van Zyl-Smit, R.N.; Campora, L.; Dezutter, N.; et al. Respiratory Syncytial Virus Prefusion F Protein Vaccine in Older Adults. N. Engl. J. Med. 2023, 388, 595–608. [Google Scholar] [CrossRef]
- Gebre, M.S.; Brito, L.A.; Tostanoski, L.H.; Edwards, D.K.; Carfi, A.; Barouch, D.H. Novel approaches for vaccine development. Cell 2021, 184, 1589–1603. [Google Scholar] [CrossRef]
- Chen, G.L.; Mithani, R.; Kapoor, A.; Lu, S.; El Asmar, L.; Panozzo, C.A.; Shaw, C.A.; Stoszek, S.K.; August, A. 234. Safety and Immunogenicity of mRNA-1345, an mRNA-Based RSV Vaccine in Younger and Older Adult Cohorts: Results from a Phase 1, Randomized Clinical Trial. Open Forum Infect. Dis. 2022, 9, ofac492.312. [Google Scholar] [CrossRef]
- Rodriguez-Fernandez, R.; Mejias, A.; Ramilo, O. Monoclonal Antibodies for Prevention of Respiratory Syncytial Virus Infection. Pediatr. Infect. Dis. J. 2021, 40, S35–S39. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.S.; Castellanos, C.A.; Chen, M.; Ngwuta, J.O.; Goodwin, E.; Moin, S.M.; Mas, V.; Melero, J.A.; Wright, P.F.; Graham, B.S.; et al. Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci. Immunol. 2016, 1, eaaj1879. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated Guidance for Palivizumab Prophylaxis Among Infants and Young Children at Increased Risk of Hospitalization for Respiratory Syncytial Virus Infection. Pediatrics 2014, 134, 415–420. [Google Scholar] [CrossRef]
- Tulloh, R.M.; Feltes, T.F. The European Forum for Clinical Management: Prophylaxis against the respiratory syncytial virus in infants and young children with congenital cardiac disease. Cardiol. Young 2005, 15, 274–278. [Google Scholar] [CrossRef]
- Zhu, Q.; McLellan, J.S.; Kallewaard, N.L.; Ulbrandt, N.D.; Palaszynski, S.; Zhang, J.; Moldt, B.; Khan, A.; Svabek, C.; McAuliffe, J.M.; et al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci. Transl. Med. 2017, 9, eaaj1928. [Google Scholar] [CrossRef]
- Hammitt, L.L.; Dagan, R.; Yuan, Y.; Baca Cots, M.; Bosheva, M.; Madhi, S.A.; Muller, W.J.; Zar, H.J.; Brooks, D.; Grenham, A.; et al. Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N. Engl. J. Med. 2022, 386, 837–846. [Google Scholar] [CrossRef]
- Griffin, M.P.; Yuan, Y.; Takas, T.; Domachowske, J.B.; Madhi, S.A.; Manzoni, P.; Simoes, E.A.F.; Esser, M.T.; Khan, A.A.; Dubovsky, F.; et al. Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants. N. Engl. J. Med. 2020, 383, 415–425. [Google Scholar] [CrossRef]
- Lin, G.L.; Drysdale, S.B.; Snape, M.D.; O’Connor, D.; Brown, A.; MacIntyre-Cockett, G.; Mellado-Gomez, E.; de Cesare, M.; Bonsall, D.; Ansari, M.A.; et al. Distinct patterns of within-host virus populations between two subgroups of human respiratory syncytial virus. Nat. Commun. 2021, 12, 5125. [Google Scholar] [CrossRef]

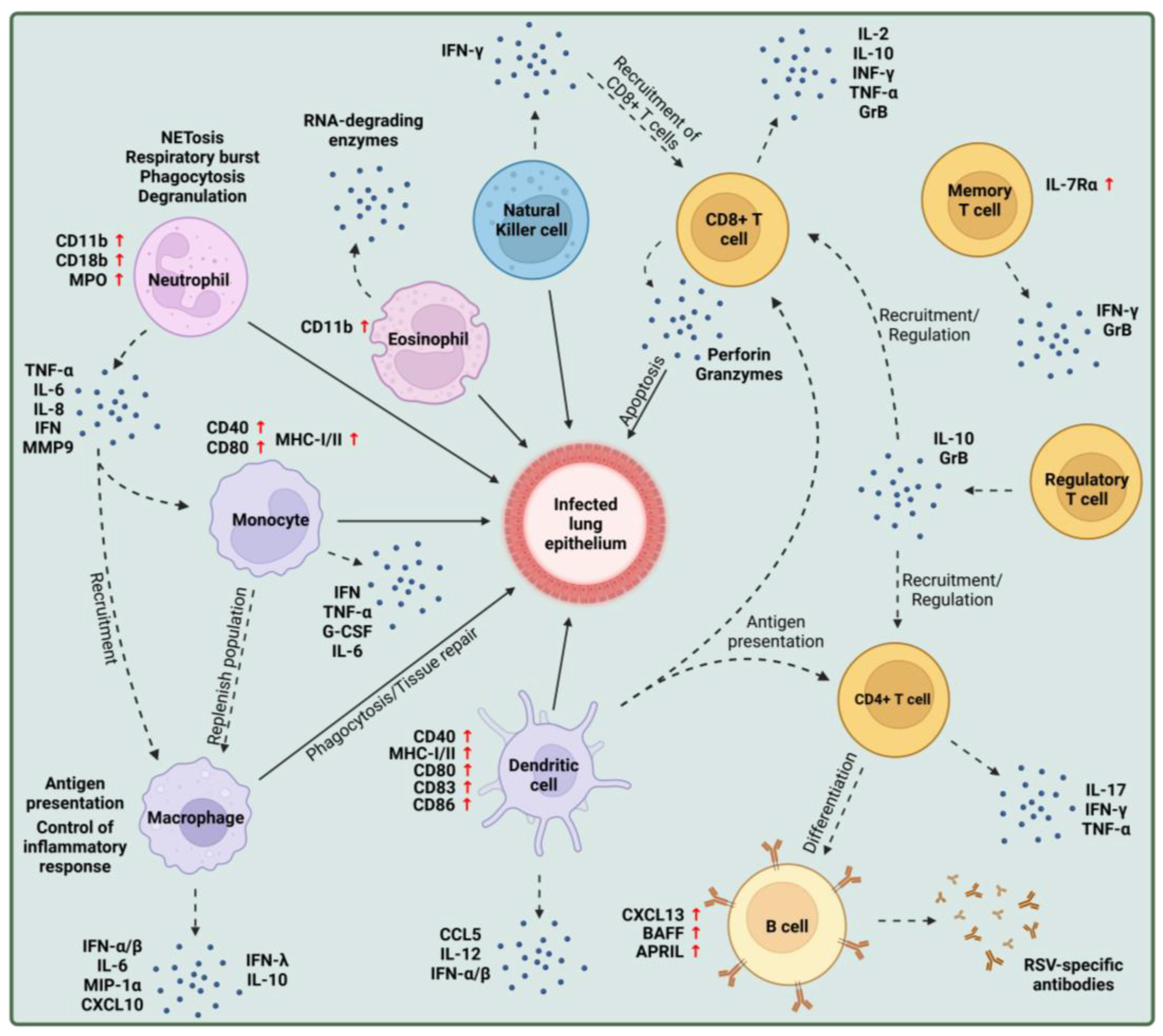


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agac, A.; Kolbe, S.M.; Ludlow, M.; Osterhaus, A.D.M.E.; Meineke, R.; Rimmelzwaan, G.F. Host Responses to Respiratory Syncytial Virus Infection. Viruses 2023, 15, 1999. https://doi.org/10.3390/v15101999
Agac A, Kolbe SM, Ludlow M, Osterhaus ADME, Meineke R, Rimmelzwaan GF. Host Responses to Respiratory Syncytial Virus Infection. Viruses. 2023; 15(10):1999. https://doi.org/10.3390/v15101999
Chicago/Turabian StyleAgac, Ayse, Sophie M. Kolbe, Martin Ludlow, Albert D. M. E. Osterhaus, Robert Meineke, and Guus F. Rimmelzwaan. 2023. "Host Responses to Respiratory Syncytial Virus Infection" Viruses 15, no. 10: 1999. https://doi.org/10.3390/v15101999
APA StyleAgac, A., Kolbe, S. M., Ludlow, M., Osterhaus, A. D. M. E., Meineke, R., & Rimmelzwaan, G. F. (2023). Host Responses to Respiratory Syncytial Virus Infection. Viruses, 15(10), 1999. https://doi.org/10.3390/v15101999