
Genomic Analysis and Taxonomic Characterization of Seven Bacteriophage Genomes Metagenomic-Assembled from the Dishui Lake
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Metagenomic Data Source
2.2. Viral Genome Assembly
2.3. Genome Characterization
2.4. Retrieval of Viruses Related to Dishui Lake Phages in Environmental Datasets
2.5. Protein-Cluster-Based Gene-Sharing Network
2.6. Proteomic Trees
2.7. Comparative Genomic Analysis
2.8. Phylogenetic Analysis
2.9. Phage Host Prediction
3. Results
3.1. Viral Community Composition in Dishui Lake
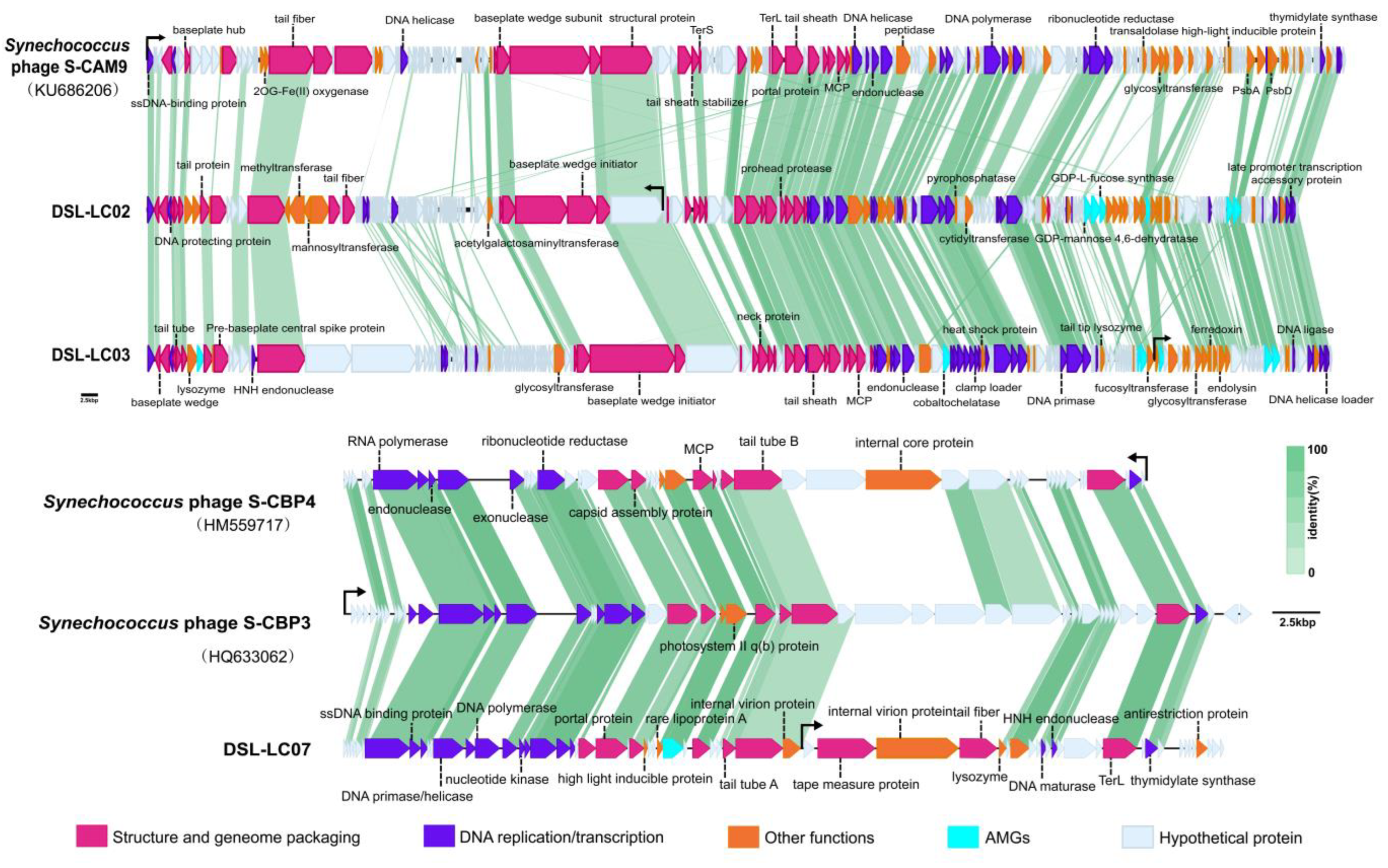
3.2. Genomic Features of Dishui Lake Phages
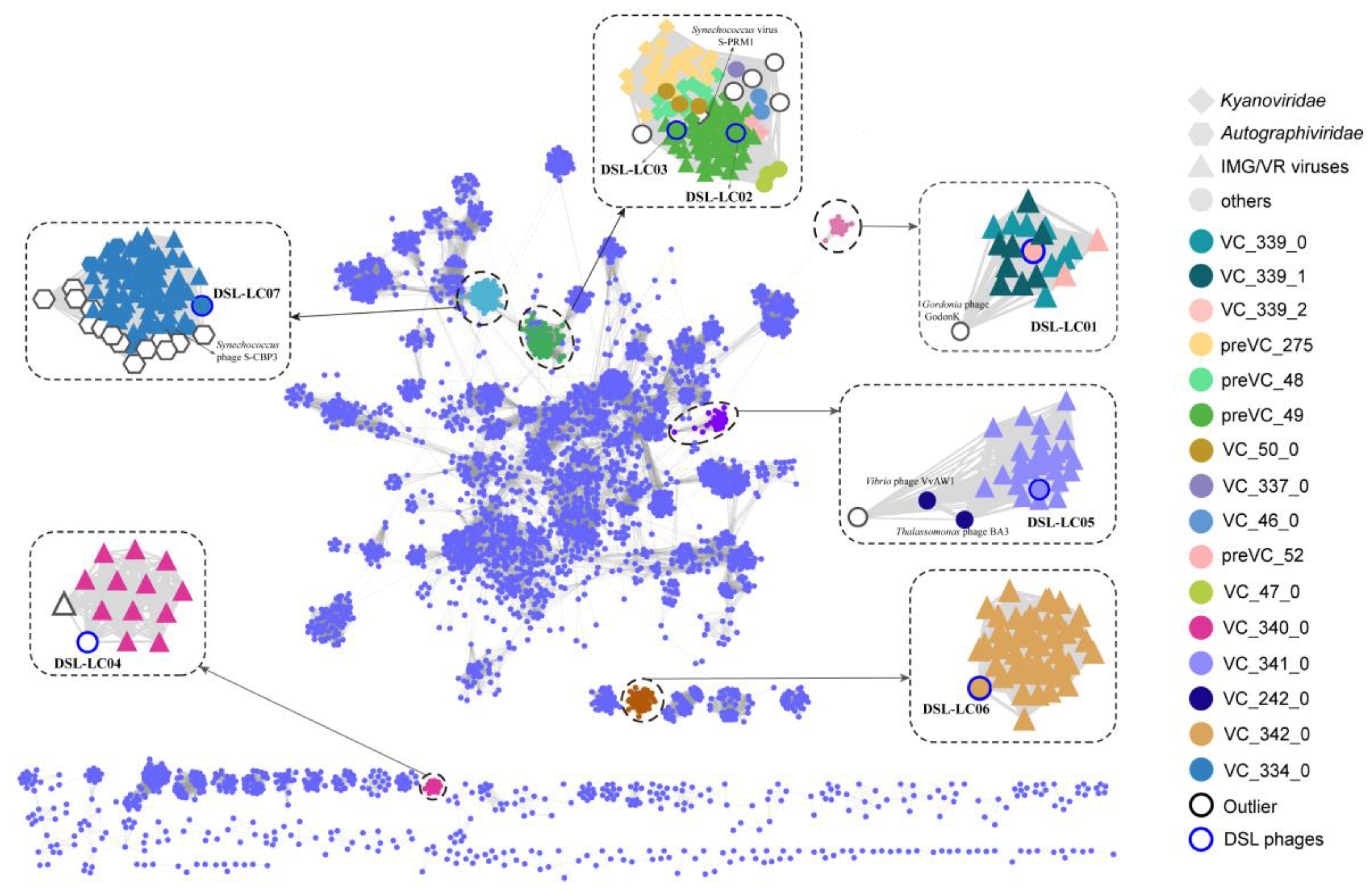
3.3. Protein-Cluster-Based Gene-Sharing Network
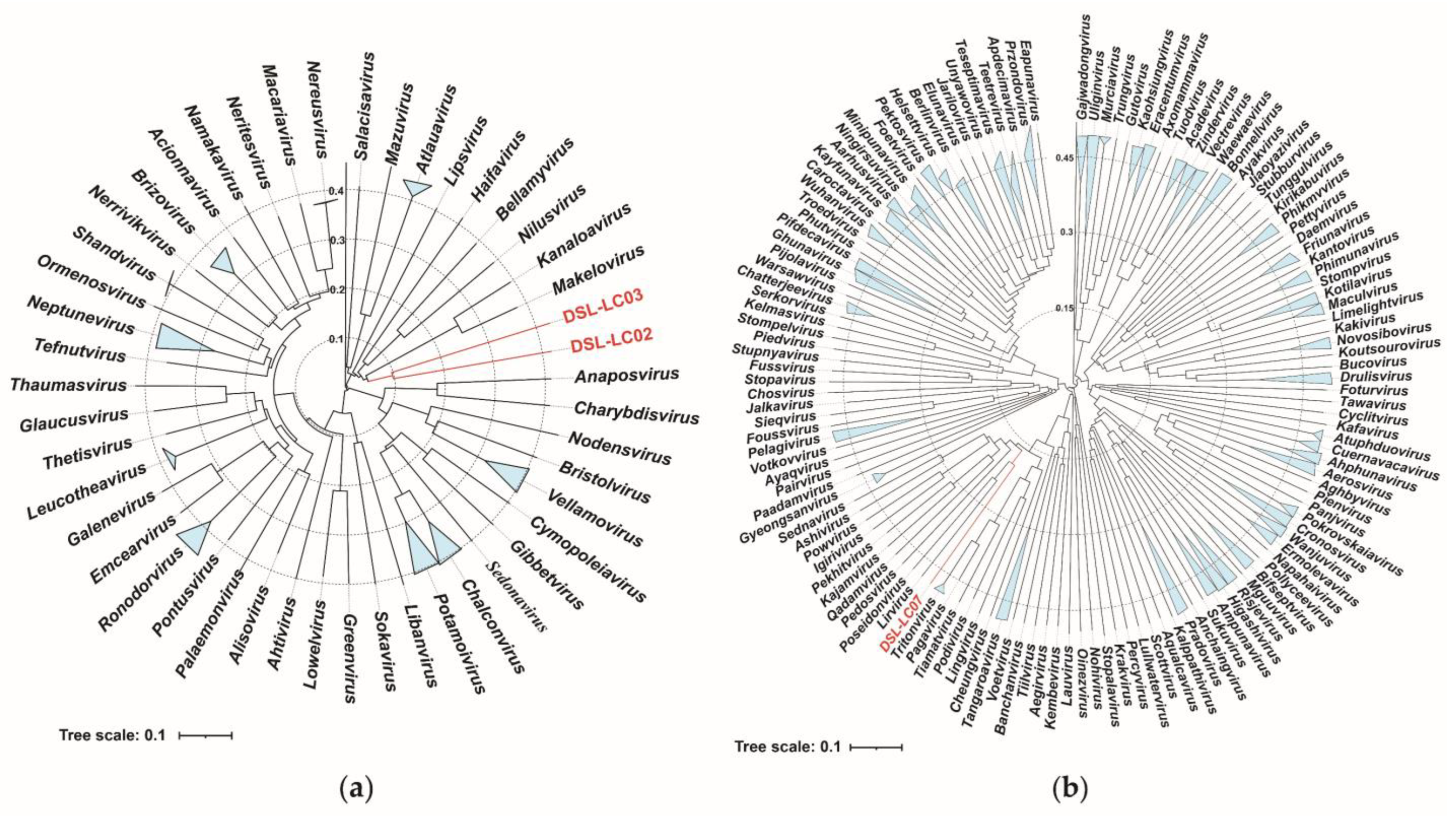
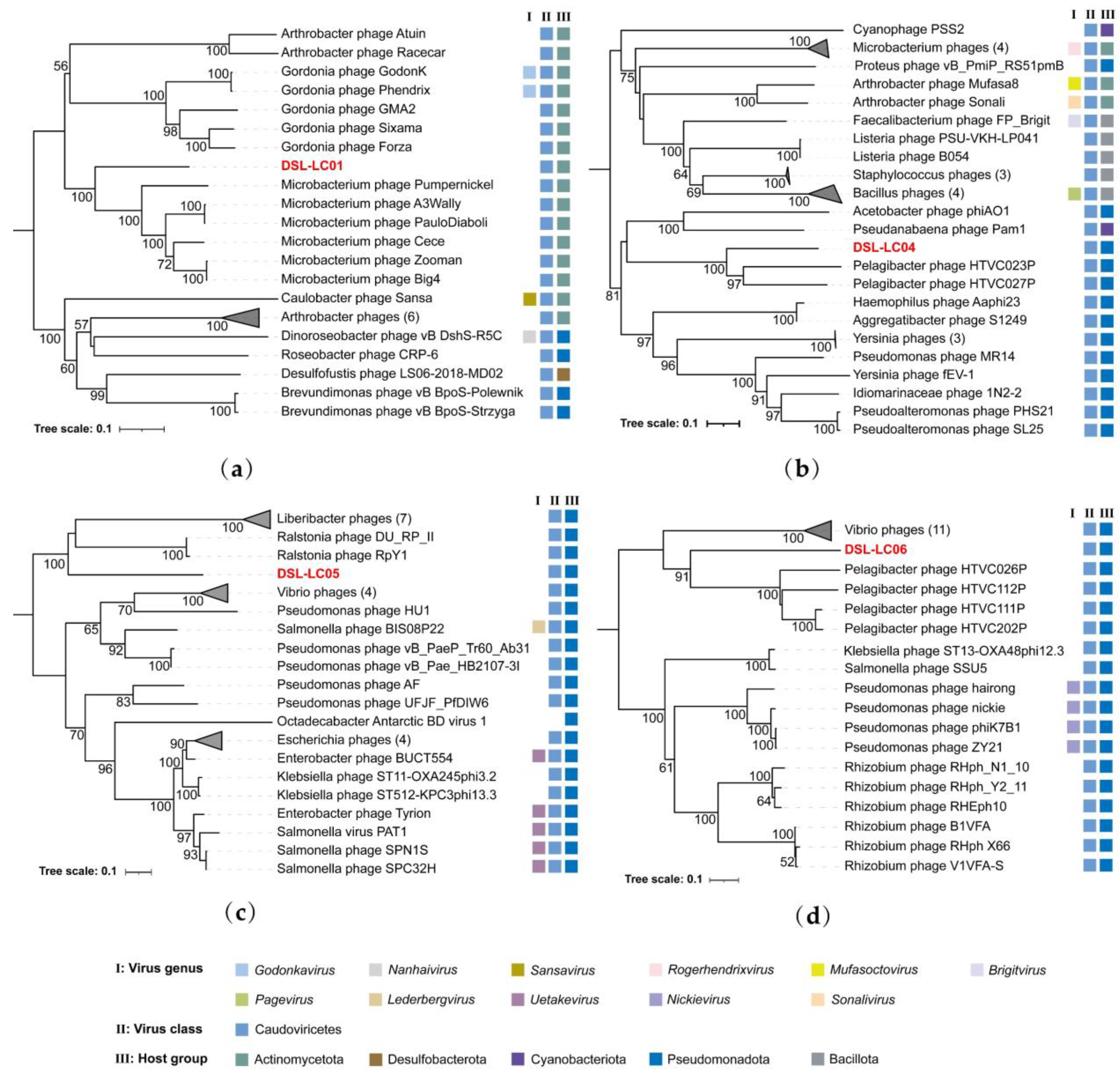
3.4. Proteomic Trees
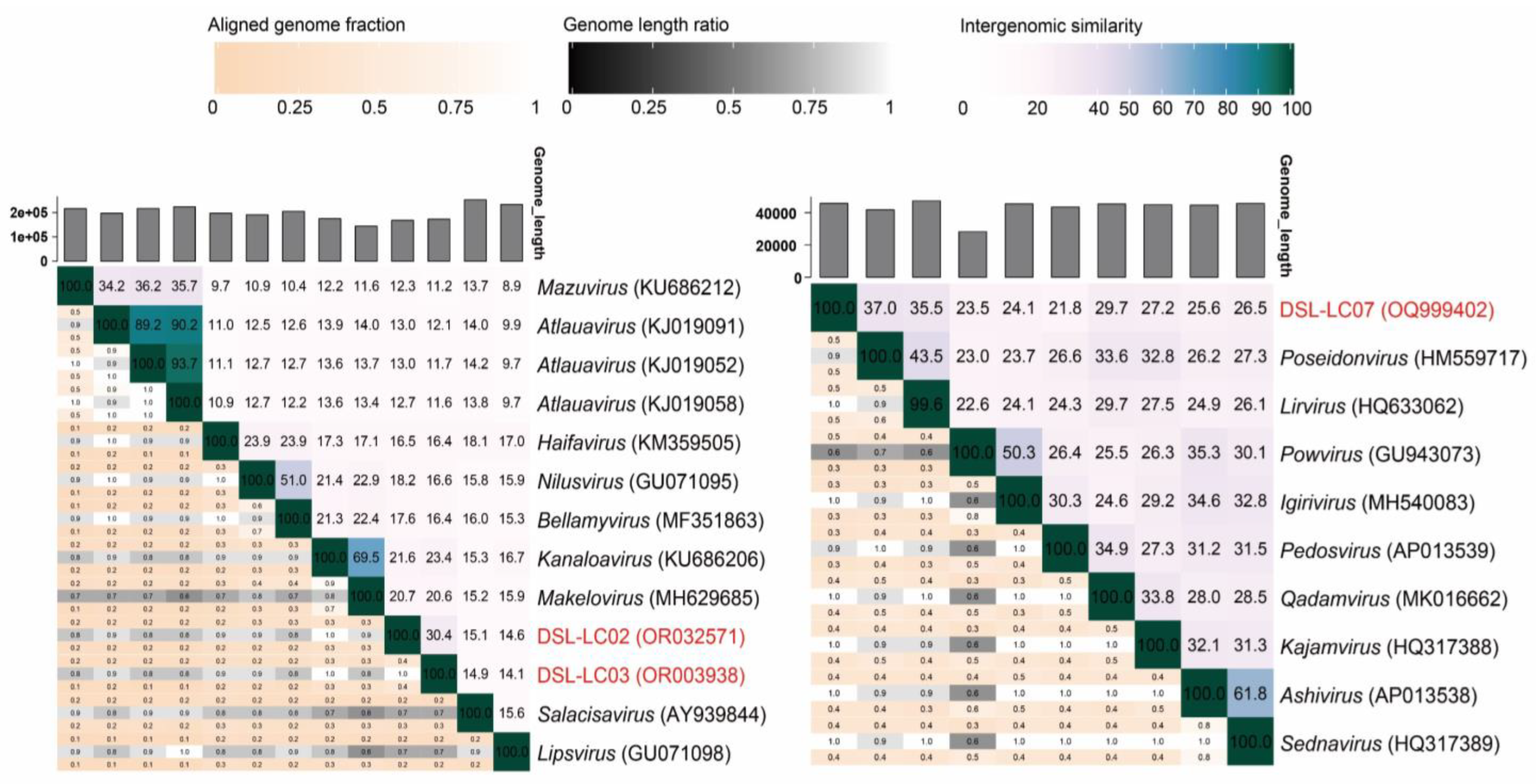
3.5. Orthologous Fraction and Nucleotide Identity of Viral Genomes
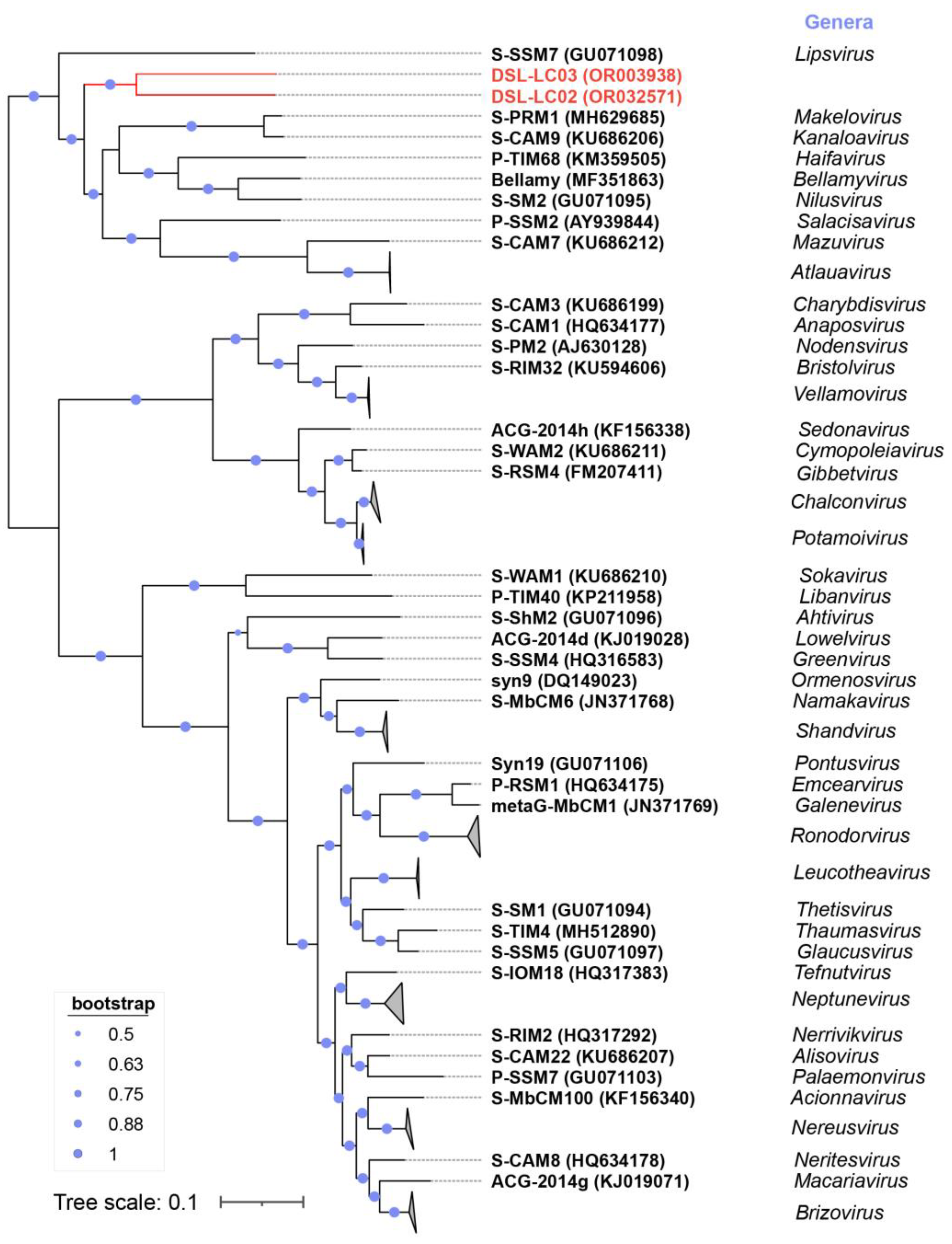
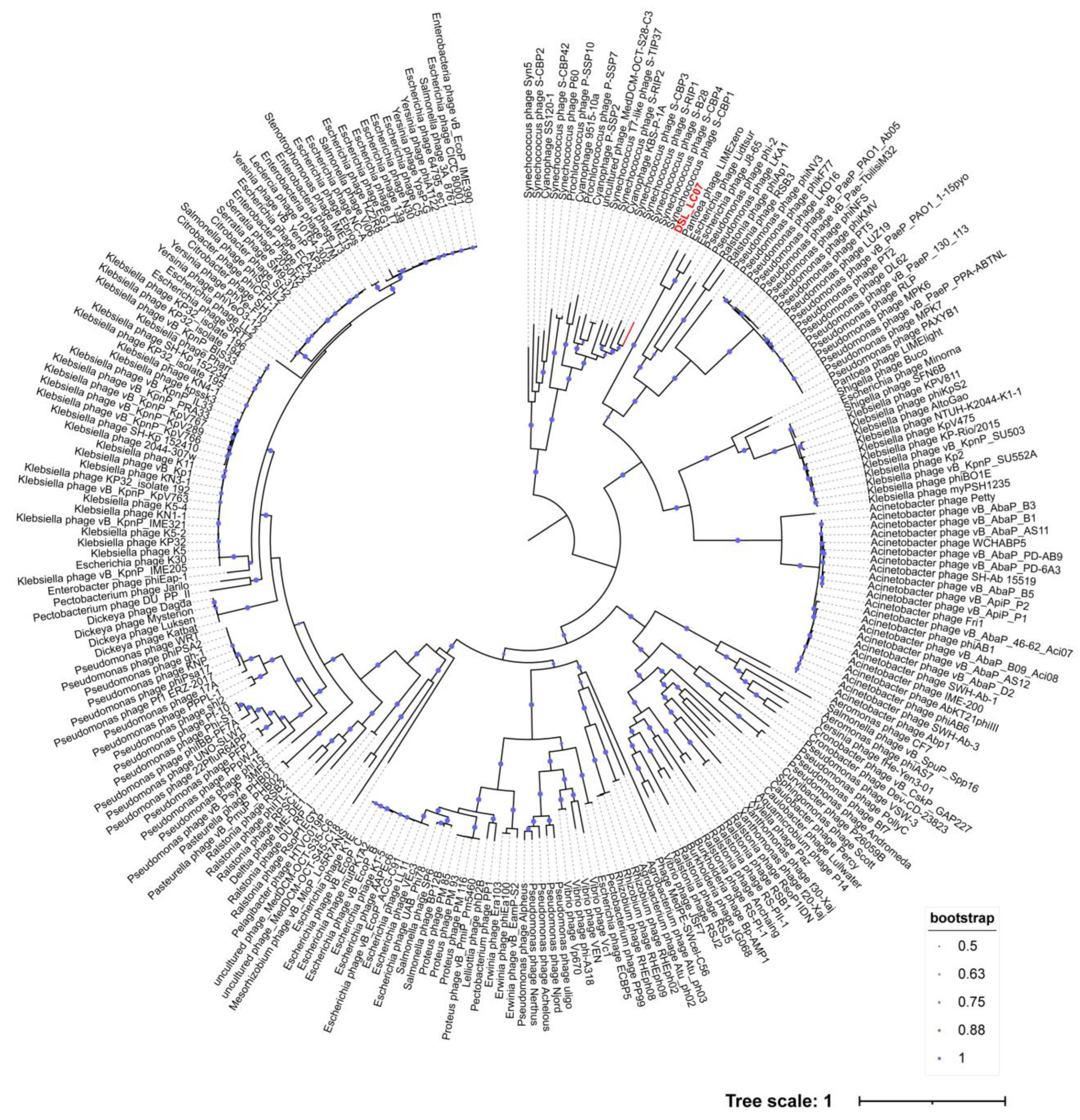
3.6. Phylogenetic Analyses
3.7. Predicted Viral Hosts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Oh, H.M.; Kang, D.; Cho, J.C. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12343–12348. [Google Scholar] [CrossRef]
- Bruder, K.; Malki, K.; Cooper, A.; Sible, E.; Shapiro, J.W.; Watkins, S.C.; Putonti, C. Freshwater Metaviromics and Bacteriophages: A Current Assessment of the State of the Art in Relation to Bioinformatic Challenges. Evol. Bioinform. Online 2016, 12, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Elbehery, A.H.A.; Deng, L. Insights into the global freshwater virome. Front. Microbiol. 2022, 13, 953500. [Google Scholar] [CrossRef] [PubMed]
- Aguirre de Carcer, D.; Lopez-Bueno, A.; Pearce, D.A.; Alcami, A. Biodiversity and distribution of polar freshwater DNA viruses. Sci. Adv. 2015, 1, e1400127. [Google Scholar] [CrossRef] [PubMed]
- Palermo, C.N.; Fulthorpe, R.R.; Saati, R.; Short, S.M. Metagenomic Analysis of Virus Diversity and Relative Abundance in a Eutrophic Freshwater Harbour. Viruses 2019, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, T.; de Leeuwe, C.; Quinn, J.P.; McGrath, J.W.; Allen, C.C.; McElarney, Y.; Watson, C.; Arkhipova, K.; Lavigne, R.; Kulakov, L.A. Metagenomic Characterisation of the Viral Community of Lough Neagh, the Largest Freshwater Lake in Ireland. PLoS ONE 2016, 11, e0150361. [Google Scholar] [CrossRef]
- Garza, D.R.; Dutilh, B.E. From cultured to uncultured genome sequences: Metagenomics and modeling microbial ecosystems. Cell Mol. Life Sci. 2015, 72, 4287–4308. [Google Scholar] [CrossRef]
- Okazaki, Y.; Nishimura, Y.; Yoshida, T.; Ogata, H.; Nakano, S.I. Genome-resolved viral and cellular metagenomes revealed potential key virus-host interactions in a deep freshwater lake. Environ. Microbiol. 2019, 21, 4740–4754. [Google Scholar] [CrossRef]
- Lu, J.; Yang, S.; Zhang, X.; Tang, X.; Zhang, J.; Wang, X.; Wang, H.; Shen, Q.; Zhang, W. Metagenomic analysis of viral community in the Yangtze River expands known eukaryotic and prokaryotic virus diversity in freshwater. Virol. Sin. 2022, 37, 60–69. [Google Scholar] [CrossRef]
- Moon, K.; Kim, S.; Kang, I.; Cho, J.C. Viral metagenomes of Lake Soyang, the largest freshwater lake in South Korea. Sci. Data 2020, 7, 349. [Google Scholar] [CrossRef] [PubMed]
- Prado, T.; Brandao, M.L.; Fumian, T.M.; Freitas, L.; Chame, M.; Leomil, L.; Magalhaes, M.G.P.; Degrave, W.M.S.; Leite, J.P.G.; Miagostovich, M.P. Virome analysis in lakes of the South Shetland Islands, Antarctica—2020. Sci. Total Environ. 2022, 852, 158537. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Yang, F.; Zhang, J.T.; Yu, R.C.; Deng, Z.; Li, W.F.; Chen, Y.; Li, Q.; Zhou, C.Z. Comparative genomic analysis of five freshwater cyanophages and reference-guided metagenomic data mining. Microbiome 2022, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Bai, M.; Xiao, W.; Zhang, S.; Wang, Y.; Cui, X. Nutrient levels and prokaryotes affect viral communities in plateau lakes. Sci. Total Environ. 2022, 839, 156033. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, P.G.; Pipas, J.M. Detecting viral sequences in NGS data. Curr. Opin. Virol. 2019, 39, 41–48. [Google Scholar] [CrossRef]
- Andrade-Martinez, J.S.; Camelo Valera, L.C.; Chica Cardenas, L.A.; Forero-Junco, L.; Lopez-Leal, G.; Moreno-Gallego, J.L.; Rangel-Pineros, G.; Reyes, A. Computational Tools for the Analysis of Uncultivated Phage Genomes. Microbiol. Mol. Biol. Rev. 2022, 86, e0000421. [Google Scholar] [CrossRef]
- Chibani, C.M.; Farr, A.; Klama, S.; Dietrich, S.; Liesegang, H. Classifying the Unclassified: A Phage Classification Method. Viruses 2019, 11, 195. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ding, C.; Achal, V.; Shan, D.; Zhou, Y.; Xu, Y.; Xiang, W.N. Potential for nutrient removal by integrated remediation methods in a eutrophicated artificial lake—A case study in Dishui Lake, Lingang New City, China. Water Sci. Technol. 2014, 70, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tan, W.; Wu, X.; Wu, Z.; Yu, G.; Li, R. First report of microcystin production in Microcystis smithii Komarek and Anagnostidis (Cyanobacteria) from a water bloom in Eastern China. J. Environ. Sci. 2011, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Cao, Y.; Pang, W.T.; Wang, L.Z.; Song, K.; You, Q.M.; Wang, Q.X. Long-term plankton community dynamics and influencing factors in a man-made shallow lake, Lake Dishui, China. Aquat. Sci. 2020, 83, 210. [Google Scholar] [CrossRef]
- Zhu, W.J.; Pan, Y.D.; Tao, J.J.; Li, X.B.; Xu, X.L.; Wang, Y.F.; Wang, Q.X. Phytoplankton community and succession in a newly man-made shallow lake, Shanghai, China. Aquat. Ecol. 2013, 47, 137–147. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Li, X.; Pan, Y.; Yan, S.; Wang, Y. The genome of a prasinoviruses-related freshwater virus reveals unusual diversity of phycodnaviruses. BMC Genom. 2018, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Zhang, W.; Zhou, X.; Wang, H.; Sun, G.; Xiao, J.; Pan, Y.; Yan, S.; Wang, Y. Novel Virophages Discovered in a Freshwater Lake in China. Front. Microbiol. 2016, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Wu, Z.; Xu, S.; Wang, Y. Isolation and Identification of a Large Green Alga Virus (Chlorella Virus XW01) of Mimiviridae and Its Virophage (Chlorella Virus Virophage SW01) by Using Unicellular Green Algal Cultures. J. Virol. 2022, 96, e0211421. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, L.; Liang, X.; Zhou, Y.; Chen, H.; Yan, S.; Wang, Y. Novel Cell-Virus-Virophage Tripartite Infection Systems Discovered in the Freshwater Lake Dishui Lake in Shanghai, China. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Tisza, M.J.; Belford, A.K.; Dominguez-Huerta, G.; Bolduc, B.; Buck, C.B. Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol. 2021, 7, veaa100. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Kurgan, L. Sequence Similarity Searching. Curr. Protoc. Protein Sci. 2019, 95, e71. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Soding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.C.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting bacteriophage lifestyle from conserved protein domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Camargo, A.P.; Nayfach, S.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 2022, 51, D733–D743. [Google Scholar] [CrossRef]
- Bartlau, N.; Wichels, A.; Krohne, G.; Adriaenssens, E.M.; Heins, A.; Fuchs, B.M.; Amann, R.; Moraru, C. Highly diverse flavobacterial phages isolated from North Sea spring blooms. ISME J. 2022, 16, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Roux, S.; Camargo, A.P.; Coutinho, F.H.; Dabdoub, S.M.; Dutilh, B.E.; Nayfach, S.; Tritt, A. iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLoS Biol. 2023, 21, e3002083. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Sun, Y. Predicting the hosts of prokaryotic viruses using GCN-based semi-supervised learning. BMC Biol. 2021, 19, 250. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.B.; Plante, P.L.; Zufferey, E.; Shah, S.A.; Corbeil, J.; Moineau, S. Streamlining CRISPR spacer-based bacterial host predictions to decipher the viral dark matter. Nucleic Acids Res. 2021, 49, 3127–3138. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, L.; Yan, S.; Chen, L.; Krupovic, M.; Wang, Y. Diverse viruses of marine archaea discovered using metagenomics. Environ. Microbiol. 2023, 25, 367–382. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. The LUCA and its complex virome. Nat. Rev. Microbiol. 2020, 18, 661–670. [Google Scholar] [CrossRef]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Lobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef]
- Bi, L.; Yu, D.T.; Du, S.; Zhang, L.M.; Zhang, L.Y.; Wu, C.F.; Xiong, C.; Han, L.L.; He, J.Z. Diversity and potential biogeochemical impacts of viruses in bulk and rhizosphere soils. Environ. Microbiol. 2021, 23, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, F.; Chen, F.; Chu, X.; Luo, H.; Zhang, R.; Du, S.; Tian, Z.; Zhao, Y. Culturing novel and abundant pelagiphages in the ocean. Environ. Microbiol. 2021, 23, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Rusinol, M.; Martinez-Puchol, S.; Timoneda, N.; Fernandez-Cassi, X.; Perez-Cataluna, A.; Fernandez-Bravo, A.; Moreno-Mesonero, L.; Moreno, Y.; Alonso, J.L.; Figueras, M.J.; et al. Metagenomic analysis of viruses, bacteria and protozoa in irrigation water. Int. J. Hyg. Environ. Health 2020, 224, 113440. [Google Scholar] [CrossRef]
- Somerville, V.; Lutz, S.; Schmid, M.; Frei, D.; Moser, A.; Irmler, S.; Frey, J.E.; Ahrens, C.H. Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiol. 2019, 19, 143. [Google Scholar] [CrossRef] [PubMed]
- Sulcius, S.; Simoliunas, E.; Alzbutas, G.; Gasiunas, G.; Jauniskis, V.; Kuznecova, J.; Miettinen, S.; Nilsson, E.; Meskys, R.; Roine, E.; et al. Genomic Characterization of Cyanophage vB_AphaS-CL131 Infecting Filamentous Diazotrophic Cyanobacterium Aphanizomenon flos-aquae Reveals Novel Insights into Virus-Bacterium Interactions. Appl. Environ. Microbiol. 2019, 85, e01311-18. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Smith, C.A.; Buchko, G.W.; Blaby, I.K.; Paez-Espino, D.; Kyrpides, N.C.; Yoshikuni, Y.; McDermott, J.E.; Hofmockel, K.S.; Cort, J.R.; et al. Structural characterization of a soil viral auxiliary metabolic gene product—A functional chitosanase. Nat. Commun. 2022, 13, 5485. [Google Scholar] [CrossRef]
- Luo, X.Q.; Wang, P.; Li, J.L.; Ahmad, M.; Duan, L.; Yin, L.Z.; Deng, Q.Q.; Fang, B.Z.; Li, S.H.; Li, W.J. Viral community-wide auxiliary metabolic genes differ by lifestyles, habitats, and hosts. Microbiome 2022, 10, 190. [Google Scholar] [CrossRef]
- Reynolds, P.E. Control of peptidoglycan synthesis in vancomycin-resistant enterococci: D,D-peptidases and D,D-carboxypeptidases. Cell Mol. Life Sci. 1998, 54, 325–331. [Google Scholar] [CrossRef]
- Tanner, J.J.; Boechi, L.; Andrew McCammon, J.; Sobrado, P. Structure, mechanism, and dynamics of UDP-galactopyranose mutase. Arch. Biochem. Biophys. 2014, 544, 128–141. [Google Scholar] [CrossRef]
- Bochow, S.; Elliman, J.; Owens, L. Bacteriophage adenine methyltransferase: A life cycle regulator? Modelled using Vibrio harveyi myovirus like. J. Appl. Microbiol. 2012, 113, 1001–1013. [Google Scholar] [CrossRef]
- van Pee, K.H.; Patallo, E.P. Flavin-dependent halogenases involved in secondary metabolism in bacteria. Appl. Microbiol. Biotechnol. 2006, 70, 631–641. [Google Scholar] [CrossRef]
- Rybniker, J.; Nowag, A.; van Gumpel, E.; Nissen, N.; Robinson, N.; Plum, G.; Hartmann, P. Insights into the function of the WhiB-like protein of mycobacteriophage TM4-a transcriptional inhibitor of WhiB2. Mol. Microbiol. 2010, 77, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Hardy, A.; Luthe, T.; Frunzke, J. Phylogenetic Distribution of WhiB- and Lsr2-Type Regulators in Actinobacteriophage Genomes. Microbiol. Spectr. 2021, 9, e0072721. [Google Scholar] [CrossRef] [PubMed]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar] [CrossRef]
- Markine-Goriaynoff, N.; Gillet, L.; Van Etten, J.L.; Korres, H.; Verma, N.; Vanderplasschen, A. Glycosyltransferases encoded by viruses. J. Gen. Virol. 2004, 85, 2741–2754. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, F.; Wen, Y.; Hu, Y.; Yuan, Y.; Wei, M.; Zhou, Y. Cell-free enzymatic synthesis of GDP-L-fucose from mannose. AMB Express 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Coleman, M.L.; Weigele, P.; Rohwer, F.; Chisholm, S.W. Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLoS Biol. 2005, 3, e144. [Google Scholar] [CrossRef]
- Van Staalduinen, L.M.; Jia, Z.C. Post-translational hydroxylation by 2OG/Fe(ll)-dependent oxygenases as a novel regulatory mechanism in bacteria. Front. Microbiol. 2015, 5, 798. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef]
- Wang, Q.; Cai, L.; Zhang, R.; Wei, S.; Li, F.; Liu, Y.; Xu, Y. A Unique Set of Auxiliary Metabolic Genes Found in an Isolated Cyanophage Sheds New Light on Marine Phage-Host Interactions. Microbiol. Spectr. 2022, 10, e0236722. [Google Scholar] [CrossRef]
- Wang, I.N.; Smith, D.L.; Young, R. Holins: The protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef] [PubMed]
- Piacente, F.; Bernardi, C.; Marin, M.; Blanc, G.; Abergel, C.; Tonetti, M.G. Characterization of a UDP-N-acetylglucosamine biosynthetic pathway encoded by the giant DNA virus Mimivirus. Glycobiology 2014, 24, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Morgado, S.; Vicente, A.C. Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes. Viruses 2019, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Zwart, G.; Crump, B.C.; Agterveld, M.P.K.V.; Hagen, F.; Han, S.K. Typical freshwater bacteria: An analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 2002, 28, 141–155. [Google Scholar] [CrossRef]
- Cai, F.; Axen, S.D.; Kerfeld, C.A. Evidence for the widespread distribution of CRISPR-Cas system in the Phylum Cyanobacteria. RNA Biol. 2013, 10, 687–693. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef]
- Burstein, D.; Sun, C.L.; Brown, C.T.; Sharon, I.; Anantharaman, K.; Probst, A.J.; Thomas, B.C.; Banfield, J.F. Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun. 2016, 7, 10613. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Genome (bp) | %GC | No. of ORF | Annotated ORF | AMGs | tRNA | Lifestyle a |
|---|---|---|---|---|---|---|---|
| Ρ | 129,469 | 53.6 | 152 | 57 | 4 | 3 | Temperate |
| DSL-LC02 | 167,928 | 36.9 | 226 | 102 | 6 | 11 | Virulent |
| DSL-LC03 | 172,805 | 35.8 | 210 | 95 | 7 | 11 | Virulent |
| DSL-LC04 | 61,547 | 51.3 | 86 | 35 | 0 | 0 | Virulent |
| DSL-LC05 | 37,569 | 56.6 | 59 | 31 | 0 | 0 | Virulent |
| DSL-LC06 | 29,811 | 38.2 | 44 | 25 | 1 | 0 | Virulent |
| DSL-LC07 | 45,797 | 46.1 | 64 | 32 | 1 | 0 | Virulent |
| Phage | iPhoP | hostG |
|---|---|---|
| DSL-LC01 | Actinomycetota; Actinomycetia | Actinomycetota; Actinomycetia |
| DSL-LC02 | Cyanobacteriota; Cyanobacteriia; PCC-6307; Cyanobiaceae | Cyanobacteriota |
| DSL-LC03 | Cyanobacteriota; Cyanobacteriia; PCC-6307; Cyanobiaceae; Synechococcus | Cyanobacteriota |
| DSL-LC04 | Pseudomonadota; Gammaproteobacteria | Pseudomonadota; Gammaproteobacteria; Pseudomonadales; Pseudomonadaceae; Pseudomonas |
| DSL-LC05 | Pseudomonadota; Gammaproteobacteria; | Pseudomonadota; Gammaproteobacteria; Enterobacterales |
| DSL-LC06 | Pseudomonadota | - |
| DSL-LC07 | Cyanobacteriota; Cyanobacteriia; PCC-6307; Cyanobiaceae; Synechococcus | Cyanobacteriota |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, H.; Zhou, Y.; Li, X.; Xu, T.; Ni, Y.; Wu, S.; Yu, Y.; Wang, Y. Genomic Analysis and Taxonomic Characterization of Seven Bacteriophage Genomes Metagenomic-Assembled from the Dishui Lake. Viruses 2023, 15, 2038. https://doi.org/10.3390/v15102038
Cai H, Zhou Y, Li X, Xu T, Ni Y, Wu S, Yu Y, Wang Y. Genomic Analysis and Taxonomic Characterization of Seven Bacteriophage Genomes Metagenomic-Assembled from the Dishui Lake. Viruses. 2023; 15(10):2038. https://doi.org/10.3390/v15102038
Chicago/Turabian StyleCai, Haoyun, Yifan Zhou, Xiefei Li, Tianqi Xu, Yimin Ni, Shuang Wu, Yongxin Yu, and Yongjie Wang. 2023. "Genomic Analysis and Taxonomic Characterization of Seven Bacteriophage Genomes Metagenomic-Assembled from the Dishui Lake" Viruses 15, no. 10: 2038. https://doi.org/10.3390/v15102038