

ZBP1 Drives IAV-Induced NLRP3 Inflammasome Activation and Lytic Cell Death, PANoptosis, Independent of the Necroptosis Executioner MLKL

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Primary Macrophage Differentiation and Stimulation
2.3. Virus Stock Preparation
2.4. Western Blotting
2.5. Cell Death Analysis
2.6. ELISA Analyses
2.7. Statistical Analyses
3. Results
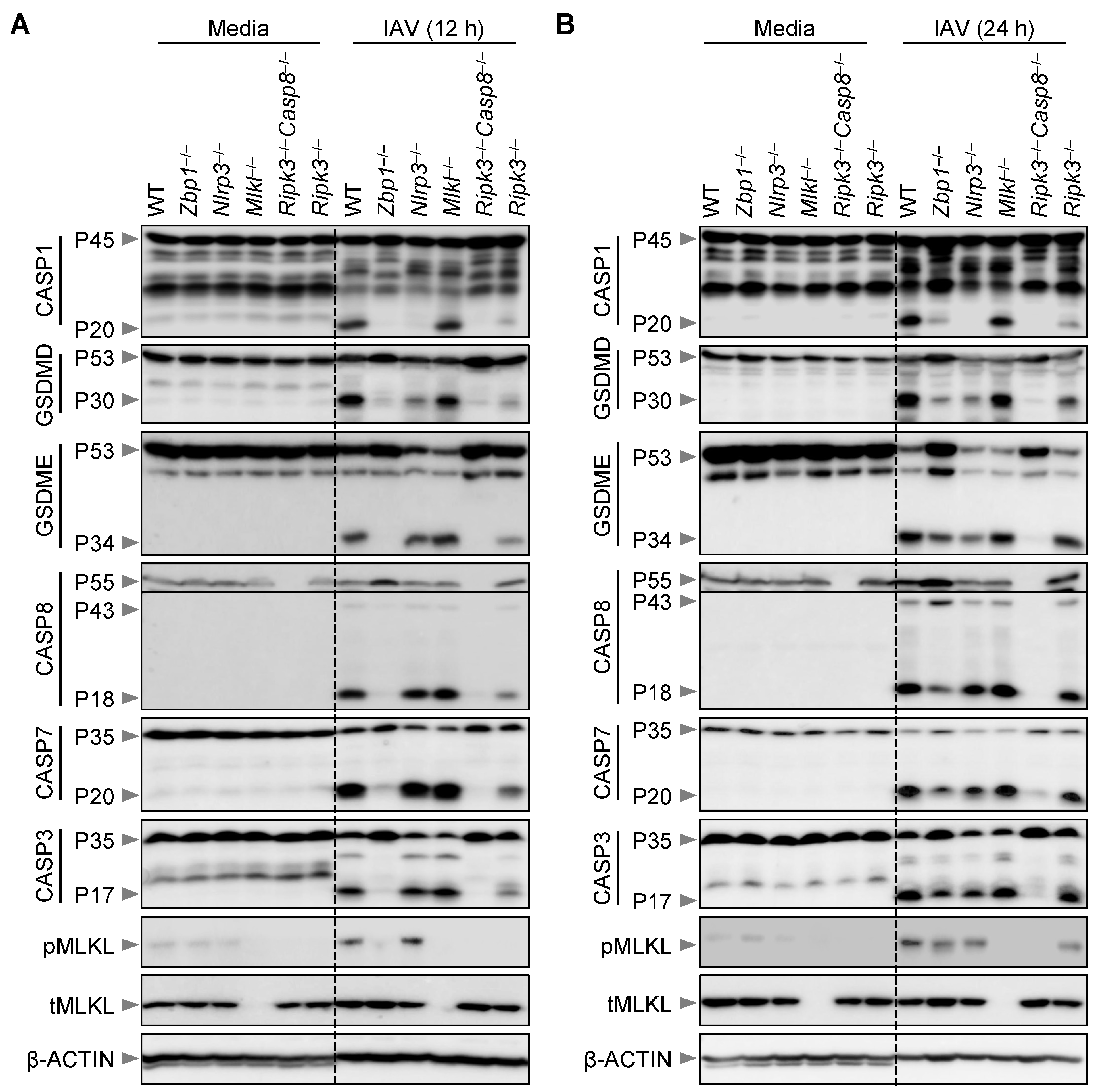
3.1. Loss of Necroptosis Executioner MLKL Does Not Protect Macrophages from IAV-Induced Cell Death
3.2. Loss of MLKL Does Not Protect Macrophages from IAV-Induced Biochemical Activation of PANoptosis
3.3. MLKL Is Not Required for IAV-Induced Inflammatory or Inflammasome-Dependent Cytokine Production
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Tumpey, T.M.; Proll, S.C.; Carter, V.; Perwitasari, O.; Thomas, M.J.; Basler, C.F.; Palese, P.; Taubenberger, J.K.; Garcia-Sastre, A.; et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 2006, 443, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Laghlali, G.; Lawlor, K.E.; Tate, M.D. Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses 2020, 12, 401. [Google Scholar] [CrossRef]
- Schulert, G.S.; Zhang, M.; Fall, N.; Husami, A.; Kissell, D.; Hanosh, A.; Zhang, K.; Davis, K.; Jentzen, J.M.; Napolitano, L.; et al. Whole-Exome Sequencing Reveals Mutations in Genes Linked to Hemophagocytic Lymphohistiocytosis and Macrophage Activation Syndrome in Fatal Cases of H1N1 Influenza. J. Infect. Dis. 2016, 213, 1180–1188. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, W.; Yang, L.; Shu, Y. The Epidemiology, Virology, and Pathogenicity of Human Infections with Avian Influenza Viruses. Cold Spring Harb. Perspect. Med. 2021, 11, a038620. [Google Scholar] [CrossRef]
- Pandian, N.; Kanneganti, T.D. PANoptosis: A unique innate immune inflammatory cell death modality. J. Immunol. 2022, 209, 1625–1633. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Kuriakose, T.; Guy, C.S.; Samir, P.; Malireddi, R.K.S.; Mishra, A.; Kanneganti, T.D. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 2017, 214, 2217–2229. [Google Scholar] [CrossRef]
- Zheng, M.; Kanneganti, T.D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687 e613. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Shubina, M.; Tummers, B.; Boyd, D.F.; Zhang, T.; Yin, C.; Gautam, A.; Guo, X.J.; Rodriguez, D.A.; Kaiser, W.J.; Vogel, P.; et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med. 2020, 217, e20191259. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129 e1113. [Google Scholar] [CrossRef]
- Lei, X.; Chen, Y.; Lien, E.; Fitzgerald, K.A. MLKL-Driven Inflammasome Activation and Caspase-8 Mediate Inflammatory Cell Death in Influenza A Virus Infection. mBio 2023, 14, e0011023. [Google Scholar] [CrossRef]
- Ishii, K.J.; Kawagoe, T.; Koyama, S.; Matsui, K.; Kumar, H.; Kawai, T.; Uematsu, S.; Takeuchi, O.; Takeshita, F.; Coban, C.; et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451, 725–729. [Google Scholar] [CrossRef]
- Newton, K.; Sun, X.; Dixit, V.M. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 2004, 24, 1464–1469. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Bynigeri, R.R.; Kancharana, B.; Sharma, B.R.; Burton, A.R.; Pelletier, S.; Kanneganti, T.D. Determining distinct roles of IL-1alpha through generation of an IL-1alpha knockout mouse with no defect in IL-1beta expression. Front. Immunol. 2022, 13, 1068230. [Google Scholar] [CrossRef]
- Tweedell, R.E.; Malireddi, R.K.S.; Kanneganti, T.D. A comprehensive guide to studying inflammasome activation and cell death. Nat. Protoc. 2020, 15, 3284–3333. [Google Scholar] [CrossRef] [PubMed]
- Tweedell, R.E.; Kanneganti, T.D. NLRP12-PANoptosome in haemolytic, infectious and inflammatory diseases. Clin. Transl. Med. 2023, 13, e1409. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 2022, 185, 3214–3231 e3223. [Google Scholar] [CrossRef]
- Denney, L.; Ho, L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Farrar, J.; Han, A.M.; Hayden, F.G.; Hyer, R.; de Jong, M.D.; Lochindarat, S.; Nguyen, T.K.; Nguyen, T.H.; Tran, T.H.; et al. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 2005, 353, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Ramos, I.; Fernandez-Sesma, A. Modulating the Innate Immune Response to Influenza A Virus: Potential Therapeutic Use of Anti-Inflammatory Drugs. Front. Immunol. 2015, 6, 361. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Craig, J.C.; Eslick, G.D.; Seppelt, I.; McLean, A.S. Use of corticosteroids in acute lung injury and acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care Med. 2009, 37, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.D.; Pickett, D.L.; van Rooijen, N.; Brooks, A.G.; Reading, P.C. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J. Virol. 2010, 84, 7569–7580. [Google Scholar] [CrossRef]
- Tate, M.D.; Schilter, H.C.; Brooks, A.G.; Reading, P.C. Responses of mouse airway epithelial cells and alveolar macrophages to virulent and avirulent strains of influenza A virus. Viral Immunol. 2011, 24, 77–88. [Google Scholar] [CrossRef]
- Taylor, R.M. Experimental Infection with Influenza a Virus in Mice: The Increase in Intrapulmonary Virus after Inoculation and the Influence of Various Factors Thereon. J. Exp. Med. 1941, 73, 43–55. [Google Scholar] [CrossRef]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef]
- Girard, M.P.; Tam, J.S.; Assossou, O.M.; Kieny, M.P. The 2009 A (H1N1) influenza virus pandemic: A review. Vaccine 2010, 28, 4895–4902. [Google Scholar] [CrossRef]
- Kandeil, A.; Patton, C.; Jones, J.C.; Jeevan, T.; Harrington, W.N.; Trifkovic, S.; Seiler, J.P.; Fabrizio, T.; Woodard, K.; Turner, J.C.; et al. Rapid evolution of A(H5N1) influenza viruses after intercontinental spread to North America. Nat. Commun. 2023, 14, 3082. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Garcia-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Gu, M.; Liu, D.; Cui, J.; Gao, G.F.; Zhou, J.; Liu, X. Epidemiology, Evolution, and Pathogenesis of H7N9 Influenza Viruses in Five Epidemic Waves since 2013 in China. Trends Microbiol. 2017, 25, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Van Kerkhove, M.D. Brief literature review for the WHO global influenza research agenda--highly pathogenic avian influenza H5N1 risk in humans. Influenza Other Respir. Viruses 2013, 7 (Suppl. 2), 26–33. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Szretter, K.J.; Katz, J.M.; Mizgerd, J.P.; Tumpey, T.M. Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J. Infect. Dis. 2010, 202, 1161–1170. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. PANoptosome signaling and therapeutic implications in infection: Central role for ZBP1 to activate the inflammasome and PANoptosis. Curr. Opin. Immunol. 2023, 83, 102348. [Google Scholar] [CrossRef]
- Karki, R.; Lee, S.; Mall, R.; Pandian, N.; Wang, Y.; Sharma, B.R.; Malireddi, R.S.; Yang, D.; Trifkovic, S.; Steele, J.A.; et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 2022, 7, eabo6294. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168 e117. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Karki, R.; Sundaram, B.; Kancharana, B.; Lee, S.; Samir, P.; Kanneganti, T.D. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons 2021, 5, 568–580. [Google Scholar] [CrossRef]
- Simpson, D.S.; Pang, J.; Weir, A.; Kong, I.Y.; Fritsch, M.; Rashidi, M.; Cooney, J.P.; Davidson, K.C.; Speir, M.; Djajawi, T.M.; et al. Interferon-gamma primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity 2022, 55, 423–441.e9. [Google Scholar] [CrossRef]
- Banoth, B.; Tuladhar, S.; Karki, R.; Sharma, B.R.; Briard, B.; Kesavardhana, S.; Burton, A.; Kanneganti, T.D. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J. Biol. Chem. 2020, 295, 18276–18283. [Google Scholar] [CrossRef]
- Cuchet-Lourenco, D.; Eletto, D.; Wu, C.; Plagnol, V.; Papapietro, O.; Curtis, J.; Ceron-Gutierrez, L.; Bacon, C.M.; Hackett, S.; Alsaleem, B.; et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 2018, 361, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Wachsmuth, L.; Kumari, S.; Schwarzer, R.; Lin, J.; Eren, R.O.; Fisher, A.; Lane, R.; Young, G.R.; Kassiotis, G.; et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 2020, 580, 391–395. [Google Scholar] [CrossRef]
- Lehle, A.S.; Farin, H.F.; Marquardt, B.; Michels, B.E.; Magg, T.; Li, Y.; Liu, Y.; Ghalandary, M.; Lammens, K.; Hollizeck, S.; et al. Intestinal Inflammation and Dysregulated Immunity in Patients With Inherited Caspase-8 Deficiency. Gastroenterology 2019, 156, 275–278. [Google Scholar] [CrossRef]
- Li, Y.; Fuhrer, M.; Bahrami, E.; Socha, P.; Klaudel-Dreszler, M.; Bouzidi, A.; Liu, Y.; Lehle, A.S.; Magg, T.; Hollizeck, S.; et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Strasser, A.; Pham, V.C.; Lill, J.R.; Roose-Girma, M.; Warming, S.; Solon, M.; et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540, 129–133. [Google Scholar] [CrossRef]
- Polykratis, A.; Martens, A.; Eren, R.O.; Shirasaki, Y.; Yamagishi, M.; Yamaguchi, Y.; Uemura, S.; Miura, M.; Holzmann, B.; Kollias, G.; et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 2019, 21, 731–742. [Google Scholar] [CrossRef]
- Schwarzer, R.; Jiao, H.; Wachsmuth, L.; Tresch, A.; Pasparakis, M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 2020, 52, 978–993 e976. [Google Scholar] [CrossRef]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.H.; et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malireddi, R.K.S.; Sharma, B.R.; Bynigeri, R.R.; Wang, Y.; Lu, J.; Kanneganti, T.-D. ZBP1 Drives IAV-Induced NLRP3 Inflammasome Activation and Lytic Cell Death, PANoptosis, Independent of the Necroptosis Executioner MLKL. Viruses 2023, 15, 2141. https://doi.org/10.3390/v15112141
Malireddi RKS, Sharma BR, Bynigeri RR, Wang Y, Lu J, Kanneganti T-D. ZBP1 Drives IAV-Induced NLRP3 Inflammasome Activation and Lytic Cell Death, PANoptosis, Independent of the Necroptosis Executioner MLKL. Viruses. 2023; 15(11):2141. https://doi.org/10.3390/v15112141
Chicago/Turabian StyleMalireddi, R. K. Subbarao, Bhesh Raj Sharma, Ratnakar R. Bynigeri, Yaqiu Wang, Jianlin Lu, and Thirumala-Devi Kanneganti. 2023. "ZBP1 Drives IAV-Induced NLRP3 Inflammasome Activation and Lytic Cell Death, PANoptosis, Independent of the Necroptosis Executioner MLKL" Viruses 15, no. 11: 2141. https://doi.org/10.3390/v15112141
APA StyleMalireddi, R. K. S., Sharma, B. R., Bynigeri, R. R., Wang, Y., Lu, J., & Kanneganti, T.-D. (2023). ZBP1 Drives IAV-Induced NLRP3 Inflammasome Activation and Lytic Cell Death, PANoptosis, Independent of the Necroptosis Executioner MLKL. Viruses, 15(11), 2141. https://doi.org/10.3390/v15112141