
Evolutionary Insight into the Association between New Jersey Polyomavirus and Humans

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction, PCR Screening and Sequencing
2.3. Short Read Archive Mining
2.4. Phylogenetic Analyses
3. Results
3.1. PCR Screening and Data Mining
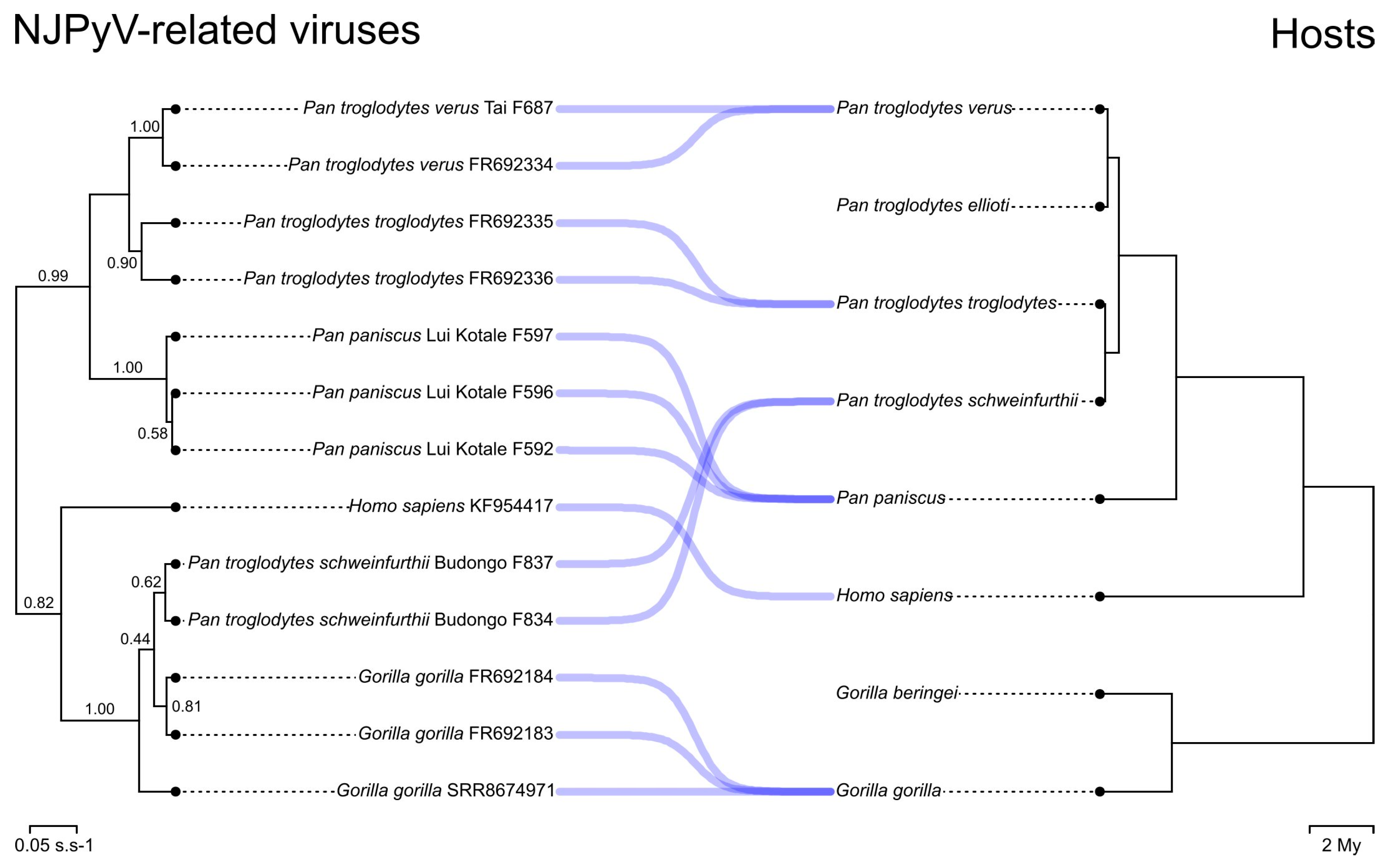
3.2. Phylogenetic Analyses
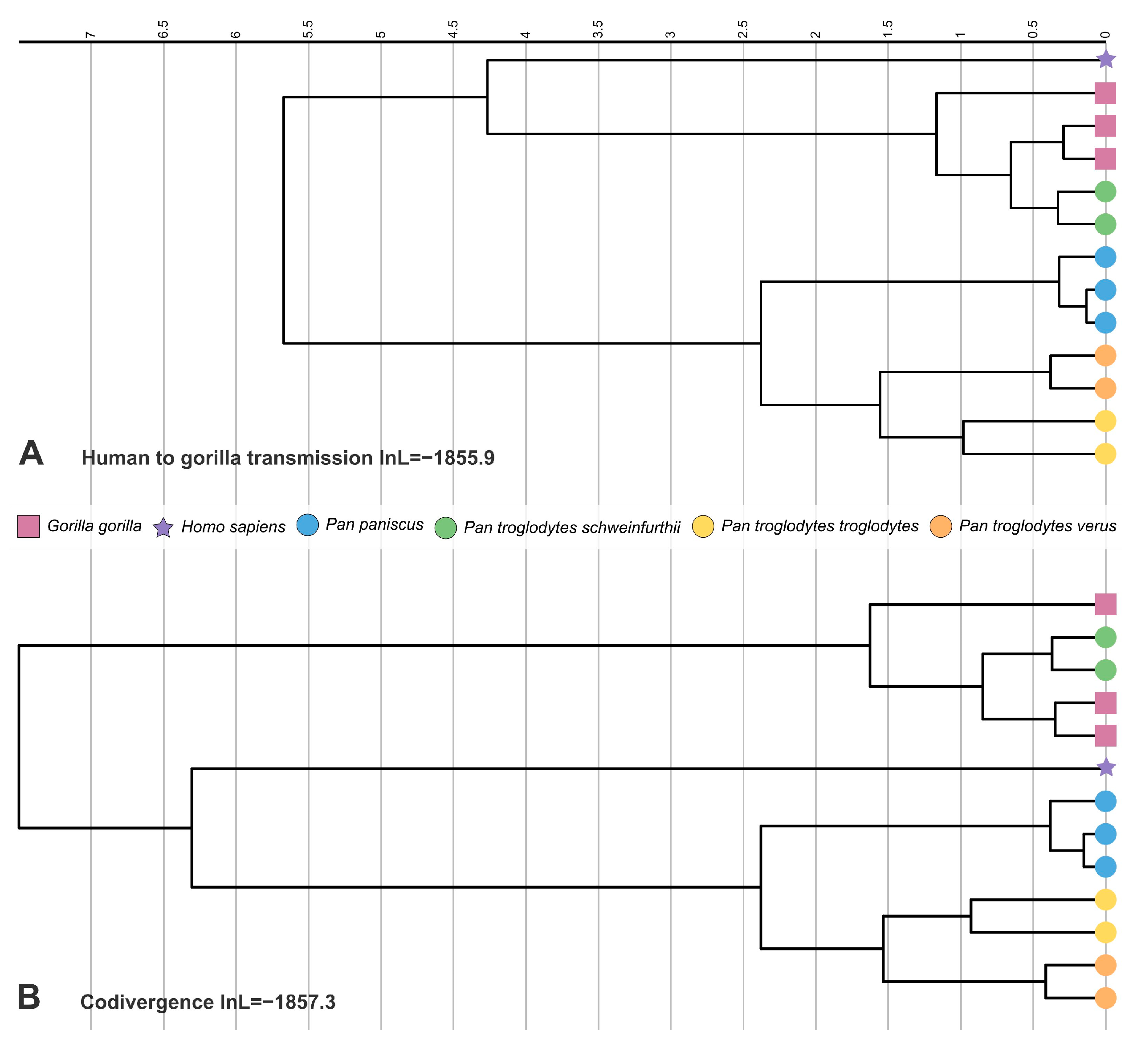
3.3. Molecular Clock Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mishra, N.; Pereira, M.; Rhodes, R.H.; An, P.; Pipas, J.M.; Jain, K.; Kapoor, A.; Briese, T.; Faust, P.L.; Lipkin, W.I. Identification of a Novel Polyomavirus in a Pancreatic Transplant Recipient with Retinal Blindness and Vasculitic Myopathy. J. Infect. Dis. 2014, 210, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Starrett, G.J.; Sappington, A.; Kostic, A.; Koren, S.; Buck, C.B.; Phillippy, A.M. Mash Screen: High-Throughput Sequence Containment Estimation for Genome Discovery. Genome Biol. 2019, 20, 232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Bai, H.; Kataoka, M.; Ito, M.; Muramatsu, M.; Suzuki, T.; Li, T.-C. Characterization of the Self-Assembly of New Jersey Polyomavirus VP1 into Virus-like Particles and the Virus Seroprevalence in Japan. Sci. Rep. 2019, 9, 13085. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, S.; van der Meijden, E.; Feltkamp, M.C.W.; Zaaijer, H.L. Seroprevalence of Fourteen Human Polyomaviruses Determined in Blood Donors. PLoS ONE 2018, 13, e0206273. [Google Scholar] [CrossRef]
- Johne, R.; Enderlein, D.; Nieper, H.; Müller, H. Novel Polyomavirus Detected in the Feces of a Chimpanzee by Nested Broad-Spectrum PCR. J. Virol. 2005, 79, 3883–3887. [Google Scholar] [CrossRef]
- Deuzing, I.; Fagrouch, Z.; Groenewoud, M.J.; Niphuis, H.; Kondova, I.; Bogers, W.; Verschoor, E.J. Detection and Characterization of Two Chimpanzee Polyomavirus Genotypes from Different Subspecies. Virol. J. 2010, 7, 347. [Google Scholar] [CrossRef]
- Camacho-Sanchez, M.; Burraco, P.; Gomez-Mestre, I.; Leonard, J.A. Preservation of RNA and DNA from Mammal Samples under Field Conditions. Mol. Ecol. Resour. 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Taylor, P.G. Reproducibility of Ancient DNA Sequences from Extinct Pleistocene Fauna. Mol. Biol. Evol. 1996, 13, 283–285. [Google Scholar] [CrossRef]
- Alves, J.M.P.; de Oliveira, A.L.; Sandberg, T.O.M.; Moreno-Gallego, J.L.; de Toledo, M.A.F.; de Moura, E.M.M.; Oliveira, L.S.; Durham, A.M.; Mehnert, D.U.; de Zanotto, P.M.A.; et al. GenSeed-HMM: A Tool for Progressive Assembly Using Profile HMMs as Seeds and Its Application in Alpavirinae Viral Discovery from Metagenomic Data. Front. Microbiol. 2016, 7, 269. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-Enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef]
- Chong, L.C.; Lauber, C. Viroid-like RNA-Dependent RNA Polymerase-Encoding Ambiviruses Are Abundant in Complex Fungi. Front. Microbiol. 2023, 14, 1144003. [Google Scholar] [CrossRef] [PubMed]
- FaBox: An Online Toolbox for Fasta Sequences-VILLESEN-2007-Molecular Ecology Notes-Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1471-8286.2007.01821.x (accessed on 31 July 2023).
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Tannier, E.; Comte, N.; Parsons, D.P. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. Methods Mol. Biol. 2021, 2231, 241–260. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessimoz, C.; Gascuel, O. Survey of Branch Support Methods Demonstrates Accuracy, Power, and Robustness of Fast Likelihood-Based Approximation Schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Ritchie, A.M.; Lo, N.; Ho, S.Y.W. The Impact of the Tree Prior on Molecular Dating of Data Sets Containing a Mixture of Inter- and Intraspecies Sampling. Syst. Biol. 2017, 66, 413–425. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Calvignac-Spencer, S.; Schulze, J.M.; Zickmann, F.; Renard, B.Y. Clock Rooting Further Demonstrates That Guinea 2014 EBOV Is a Member of the Zaïre Lineage. PLoS Curr. 2014, 6, ecurrents.outbreaks.c0e035c86d721668a6ad7353f7f6fe86. [Google Scholar] [CrossRef] [PubMed]
- Perelman, P.; Johnson, W.E.; Roos, C.; Seuánez, H.N.; Horvath, J.E.; Moreira, M.A.M.; Kessing, B.; Pontius, J.; Roelke, M.; Rumpler, Y.; et al. A Molecular Phylogeny of Living Primates. PLoS Genet. 2011, 7, e1001342. [Google Scholar] [CrossRef] [PubMed]
- Kuderna, L.F.K.; Gao, H.; Janiak, M.C.; Kuhlwilm, M.; Orkin, J.D.; Bataillon, T.; Manu, S.; Valenzuela, A.; Bergman, J.; Rousselle, M.; et al. A Global Catalog of Whole-Genome Diversity from 233 Primate Species. Science 2023, 380, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing 2023; R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 1 September 2023).
- Revell, L.J. Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- de Manuel, M.; Kuhlwilm, M.; Frandsen, P.; Sousa, V.C.; Desai, T.; Prado-Martinez, J.; Hernandez-Rodriguez, J.; Dupanloup, I.; Lao, O.; Hallast, P.; et al. Chimpanzee Genomic Diversity Reveals Ancient Admixture with Bonobos. Science 2016, 354, 477–481. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hoppe, E.; Pauly, M.; Gillespie, T.R.; Akoua-Koffi, C.; Hohmann, G.; Fruth, B.; Karhemere, S.; Madinda, N.F.; Mugisha, L.; Muyembe, J.-J.; et al. Multiple Cross-Species Transmission Events of Human Adenoviruses (HAdV) during Hominine Evolution. Mol. Biol. Evol. 2015, 32, 2072–2084. [Google Scholar] [CrossRef]
- Murthy, S.; O’Brien, K.; Agbor, A.; Angedakin, S.; Arandjelovic, M.; Ayimisin, E.A.; Bailey, E.; Bergl, R.A.; Brazzola, G.; Dieguez, P.; et al. Cytomegalovirus Distribution and Evolution in Hominines. Virus Evol. 2019, 5, vez015. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Simmonds, P. Evaluating the Evidence for Virus/Host Co-Evolution. Curr. Opin. Virol. 2011, 1, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Catacchio, C.R.; Hillier, L.W.; Porubsky, D.; Li, R.; Sulovari, A.; Fernandes, J.D.; Montinaro, F.; Gordon, D.S.; Storer, J.M.; et al. A High-Quality Bonobo Genome Refines the Analysis of Hominid Evolution. Nature 2021, 594, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The Ancient Evolutionary History of Polyomaviruses. PLoS Pathog. 2016, 12, e1005574. [Google Scholar] [CrossRef]
- Tan, Z.; Gonzalez, G.; Sheng, J.; Wu, J.; Zhang, F.; Xu, L.; Zhang, P.; Zhu, A.; Qu, Y.; Tu, C.; et al. Extensive Genetic Diversity of Polyomaviruses in Sympatric Bat Communities: Host Switching versus Coevolution. J. Virol. 2020, 94, e02101-19. [Google Scholar] [CrossRef]
- Madinda, N.F.; Ehlers, B.; Wertheim, J.O.; Akoua-Koffi, C.; Bergl, R.A.; Boesch, C.; Akonkwa, D.B.M.; Eckardt, W.; Fruth, B.; Gillespie, T.R.; et al. Assessing Host-Virus Codivergence for Close Relatives of Merkel Cell Polyomavirus Infecting African Great Apes. J. Virol. 2016, 90, 8531–8541. [Google Scholar] [CrossRef]
- Ehlers, B.; Anoh, A.E.; Ben Salem, N.; Broll, S.; Couacy-Hymann, E.; Fischer, D.; Gedvilaite, A.; Ingenhütt, N.; Liebmann, S.; Martin, M.; et al. Novel Polyomaviruses in Mammals from Multiple Orders and Reassessment of Polyomavirus Evolution and Taxonomy. Viruses 2019, 11, 930. [Google Scholar] [CrossRef]
- Vidovszky, M.Z.; Tan, Z.; Carr, M.J.; Boldogh, S.; Harrach, B.; Gonzalez, G. Bat-Borne Polyomaviruses in Europe Reveal an Evolutionary History of Intrahost Divergence with Horseshoe Bats Distributed across the African and Eurasian Continents. J. Gen. Virol. 2020, 101, 1119–1130. [Google Scholar] [CrossRef]
- Firdessa, R.; Berg, S.; Hailu, E.; Schelling, E.; Gumi, B.; Erenso, G.; Gadisa, E.; Kiros, T.; Habtamu, M.; Hussein, J.; et al. Mycobacterial Lineages Causing Pulmonary and Extrapulmonary Tuberculosis, Ethiopia. Emerg. Infect. Dis. 2013, 19, 460–463. [Google Scholar] [CrossRef]
- Burrel, S.; Boutolleau, D.; Ryu, D.; Agut, H.; Merkel, K.; Leendertz, F.H.; Calvignac-Spencer, S. Ancient Recombination Events between Human Herpes Simplex Viruses. Mol. Biol. Evol. 2017, 34, 1713–1721. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Species/Subspecies | Location (Country) | Number of Samples | Number of Positives (Number of Individuals #) |
|---|---|---|---|
| Pan troglodytes verus | Tai National Park (Côte d’Ivoire) | 153 | 4 (3) |
| Pan troglodytes troglodytes | Loango National Park (Gabon) | 24 | 0 |
| Pan troglodytes schweinfurthii | Kibale National Park (Uganda) | 70 | 0 |
| Pan troglodytes schweinfurthii | Budongo Forest (Uganda) | 3 | 2 |
| Pan paniscus | Salonga National Park (DRC *) | 10 | 3 (≥2) |
| Pan paniscus | Malebo (DRC *) | 25 | 4 |
| Gorilla beringei beringei | Bwindi Impenetrable National Park (Uganda) | 9 | 0 |
| Gorilla beringei graueri | Kahuzi-Biega National Park (DRC *) | 25 | 0 |
| Gorilla gorilla gorilla | Loango National Park (Gabon) | 10 | 0 |
| tMRCA # | Perelman et al., 2010 [23] (Mean [95%] HPD $) | Normal Prior * (mean+/−sd [Central 95%]) | Kuderna et al., 2023 [24] (Mean [95%] HPD $) | Normal Prior * (Mean+/−sd [Central 95%]) |
|---|---|---|---|---|
| Homo + Pan + Gorilla | 8.30 [6.58–10.07] | 8.30+/−0.90 [6.54–10.06] | 10.12 [8.79–11.24] | 10.12+/−0.69 [8.77–11.47] |
| Homo + Pan | 6.60 [5.40–7.96] | 6.60+/−0.70 [5.23–7.97] | 8.01 [6.91–8.96] | 8.01+/−0.56 [6.91–9.11] |
| Pan | 2.17 [1.28–3.21] | 2.17+/−0.52 [1.15–3.19] | 2.39 [1.98–2.81] | 2.39+/−0.22 [1.96–2.82] |
| Pan troglodytes | 1.24 [0.66–1.90] | 1.24+/–0.32 [0.61–1.87] | NA | NA |
| No Calibration MLE * | Calibration MLE * (2lnBFcal/no cal #) | Transmission Event Date Mean in Million Years [95% HPD &] | ||||
|---|---|---|---|---|---|---|
| Perelman et al., 2010 [23] | Kuderna et al., 2023 [24] | Perelman et al., 2010 [23] | Kuderna et al., 2023 [24] | |||
| Model | Codivergence | −1854.2 | −1857.3 (−6.2) | −1857.8 (−7.2) | NA | NA |
| Incomplete lineage sorting | −1852.3 | −1858.0 (−11.4) | −1858.6 (−12.6) | NA | NA | |
| Gorilla to human | −1857.3 (−10.0) | −1858.0 (−11.4) | 4.91 [2.37–7.57] | 6.44 [2.75–9.67] | ||
| Human to gorilla | −1855.9 (−7.2) | −1856.8 (−9.0) | 4.29 [2.40–6.07] | 5.55 [3.08–7.79] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghebatrafat, A.-A.; Lauber, C.; Merkel, K.; Fruth, B.; Langergraber, K.; Robbins, M.M.; Wittig, R.M.; Leendertz, F.H.; Calvignac-Spencer, S. Evolutionary Insight into the Association between New Jersey Polyomavirus and Humans. Viruses 2023, 15, 2248. https://doi.org/10.3390/v15112248
Aghebatrafat A-A, Lauber C, Merkel K, Fruth B, Langergraber K, Robbins MM, Wittig RM, Leendertz FH, Calvignac-Spencer S. Evolutionary Insight into the Association between New Jersey Polyomavirus and Humans. Viruses. 2023; 15(11):2248. https://doi.org/10.3390/v15112248
Chicago/Turabian StyleAghebatrafat, Aref-Abdolllah, Chris Lauber, Kevin Merkel, Barbara Fruth, Kevin Langergraber, Martha M. Robbins, Roman M. Wittig, Fabian H. Leendertz, and Sébastien Calvignac-Spencer. 2023. "Evolutionary Insight into the Association between New Jersey Polyomavirus and Humans" Viruses 15, no. 11: 2248. https://doi.org/10.3390/v15112248