
Viral Diversity in Benthic Abyssal Ecosystems: Ecological and Methodological Considerations
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sediment Collection and Viral DNA Extraction
2.2. Library Preparation and Illumina Short-Read Sequencing
2.3. Nanopore Library Preparation and Sequencing
2.4. Sequencing Data Processing
2.5. Identification and Analysis of Viral Sequences
3. Results
3.1. Comparison between Unamplified and Randomly Amplified Benthic Abyssal Viromes
3.2. Taxonomic Composition of the Benthic Abyssal Viromes
3.3. Output of the Long Read Sequecing of the Amplififed Virome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Ocean Plankton. Patterns and Ecological Drivers of Ocean Viral Communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef]
- Yoshida, M.; Mochizuki, T.; Urayama, S.-I.; Yoshida-Takashima, Y.; Nishi, S.; Hirai, M.; Nomaki, H.; Takaki, Y.; Nunoura, T.; Takai, K. Quantitative Viral Community DNA Analysis Reveals the Dominance of Single-Stranded DNA Viruses in Offshore Upper Bathyal Sediment from Tohoku, Japan. Front. Microbiol. 2018, 9, 75. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109–1123.e14. [Google Scholar] [CrossRef]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Eveillard, D.; Gorsky, G.; Guidi, L.; Iudicone, D.; Karsenti, E.; Lombard, F.; et al. Tara Oceans: Towards Global Ocean Ecosystems Biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Ke, F.; Gui, L.; Zhao, Z. Recent Insights into Aquatic Viruses: Emerging and Reemerging Pathogens, Molecular Features, Biological Effects, and Novel Investigative Approaches. Water Biol. Secur. 2022, 1, 100062. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Robertsen, E.M.; Landfald, B. Viral Assemblage Variation in an Arctic Shelf Seafloor. Aquat. Microb. Ecol. 2017, 78, 135–145. [Google Scholar] [CrossRef]
- Corinaldesi, C.; Tangherlini, M.; Dell’Anno, A. From Virus Isolation to Metagenome Generation for Investigating Viral Diversity in Deep-Sea Sediments. Sci. Rep. 2017, 7, 8355. [Google Scholar] [CrossRef]
- Yoshida, M.; Takaki, Y.; Eitoku, M.; Nunoura, T.; Takai, K. Metagenomic Analysis of Viral Communities in (Hado)Pelagic Sediments. PLoS ONE 2013, 8, e57271. [Google Scholar] [CrossRef]
- Li, Z.; Pan, D.; Wei, G.; Pi, W.; Zhang, C.; Wang, J.-H.; Peng, Y.; Zhang, L.; Wang, Y.; Hubert, C.R.J.; et al. Deep Sea Sediments Associated with Cold Seeps Are a Subsurface Reservoir of Viral Diversity. ISME J. 2021, 15, 2366–2378. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, W.; Dai, X.; Zhu, Y.; Wang, J.; Zhu, Y.; Zheng, H.; Huang, Y.; Dong, Z.; Du, W.; et al. Extraordinary Diversity of Viruses in Deep-Sea Sediments as Revealed by Metagenomics without Prior Virion Separation. Environ. Microbiol. 2021, 23, 728–743. [Google Scholar] [CrossRef]
- Zhao, J.; Jing, H.; Wang, Z.; Wang, L.; Jian, H.; Zhang, R.; Xiao, X.; Chen, F.; Jiao, N.; Zhang, Y. Novel Viral Communities Potentially Assisting in Carbon, Nitrogen, and Sulfur Metabolism in the Upper Slope Sediments of Mariana Trench. mSystems 2022, 7, e0135821. [Google Scholar] [CrossRef]
- Bäckström, D.; Yutin, N.; Jørgensen, S.L.; Dharamshi, J.; Homa, F.; Zaremba-Niedwiedzka, K.; Spang, A.; Wolf, Y.I.; Koonin, E.V.; Ettema, T.J.G. Virus Genomes from Deep Sea Sediments Expand the Ocean Megavirome and Support Independent Origins of Viral Gigantism. mBio 2019, 10, e02497-18. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, H.; Huang, Y.; Xie, Z.; Zhang, M.; Wei, Y.; Li, J.; Ma, Y.; Luo, M.; Ding, W.; et al. Revealing the Full Biosphere Structure and Versatile Metabolic Functions in the Deepest Ocean Sediment of the Challenger Deep. Genome Biol. 2021, 22, 207. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origin of Viruses: Primordial Replicators Recruiting Capsids from Hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Pan, D.; Morono, Y.; Inagaki, F.; Takai, K. An Improved Method for Extracting Viruses From Sediment: Detection of Far More Viruses in the Subseafloor Than Previously Reported. Front. Microbiol. 2019, 10, 878. [Google Scholar] [CrossRef]
- Danovaro, R.; Middelboe, M. Separation of free virus particles from sediments in aquatic systems. In Manual of Aquatic Viral Ecology; Wilhelm, S.W., Weinbauer, M.G., Suttle, C.A., Eds.; ASLO: Washington, DC, USA, 2010; pp. 74–81. [Google Scholar]
- Malki, K.; Rosario, K.; Sawaya, N.A.; Székely, A.J.; Tisza, M.J.; Breitbart, M. Prokaryotic and Viral Community Composition of Freshwater Springs in Florida, USA. mBio 2020, 11, e00436-20. [Google Scholar] [CrossRef]
- Hirai, M.; Nishi, S.; Tsuda, M.; Sunamura, M.; Takaki, Y.; Nunoura, T. Library Construction from Subnanogram DNA for Pelagic Sea Water and Deep-Sea Sediments. Microbes Environ. 2017, 32, 336–343. [Google Scholar] [CrossRef]
- Zablocki, O.; Michelsen, M.; Burris, M.; Solonenko, N.; Warwick-Dugdale, J.; Ghosh, R.; Pett-Ridge, J.; Sullivan, M.B.; Temperton, B. VirION2: A Short- and Long-Read Sequencing and Informatics Workflow to Study the Genomic Diversity of Viruses in Nature. PeerJ 2021, 9, e11088. [Google Scholar] [CrossRef]
- Willenbücher, K.; Wibberg, D.; Huang, L.; Conrady, M.; Ramm, P.; Gätcke, J.; Busche, T.; Brandt, C.; Szewzyk, U.; Schlüter, A.; et al. Phage Genome Diversity in a Biogas-Producing Microbiome Analyzed by Illumina and Nanopore GridION Sequencing. Microorganisms 2022, 10, 368. [Google Scholar] [CrossRef]
- Beaulaurier, J.; Luo, E.; Eppley, J.M.; Uyl, P.D.; Dai, X.; Burger, A.; Turner, D.J.; Pendelton, M.; Juul, S.; Harrington, E.; et al. Assembly-Free Single-Molecule Sequencing Recovers Complete Virus Genomes from Natural Microbial Communities. Genome Res. 2020, 30, 437–446. [Google Scholar] [CrossRef]
- Turnewitsch, R.; Lahajnar, N.; Haeckel, M.; Christiansen, B. An Abyssal Hill Fractionates Organic and Inorganic Matter in Deep-Sea Surface Sediments. Geophys. Res. Lett. 2015, 42, 7663–7672. [Google Scholar] [CrossRef]
- Durden, J.M.; Bett, B.J.; Jones, D.O.B.; Huvenne, V.A.I.; Ruhl, H.A. Abyssal Hills—Hidden Source of Increased Habitat Heterogeneity, Benthic Megafaunal Biomass and Diversity in the Deep Sea. Prog. Oceanogr. 2015, 137, 209–218. [Google Scholar] [CrossRef]
- Durden, J.M.; Ruhl, H.A.; Pebody, C.; Blackbird, S.J.; van Oevelen, D. Differences in the Carbon Flows in the Benthic Food Webs of Abyssal Hill and Plain Habitats. Limnol. Oceanogr. 2017, 62, 1771–1782. [Google Scholar] [CrossRef]
- Durden, J.M.; Bett, B.J.; Ruhl, H.A. Subtle Variation in Abyssal Terrain Induces Significant Change in Benthic Megafaunal Abundance, Diversity, and Community Structure. Prog. Oceanogr. 2020, 186, 102395. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate Paired Shotgun Read Merging via Overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Marijon, P.; Chikhi, R.; Varré, J.-S. Yacrd and Fpa: Upstream Tools for Long-Read Genome Assembly. Bioinformatics 2020, 36, 3894–3896. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. metaFlye: Scalable Long-Read Metagenome Assembly Using Repeat Graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef]
- Pandolfo, M.; Telatin, A.; Lazzari, G.; Adriaenssens, E.M.; Vitulo, N. MetaPhage: An Automated Pipeline for Analyzing, Annotating, and Classifying Bacteriophages in Metagenomics Sequencing Data. mSystems 2022, 7, e0074122. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining Viral Signal from Microbial Genomic Data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying Viruses from Metagenomic Data Using Deep Learning. Quant. Biol. Beijing China 2020, 8, 64–77. [Google Scholar] [CrossRef]
- Starikova, E.V.; Tikhonova, P.O.; Prianichnikov, N.A.; Rands, C.M.; Zdobnov, E.M.; Ilina, E.N.; Govorun, V.M. Phigaro: High-Throughput Prophage Sequence Annotation. Bioinformatics 2020, 36, 3882–3884. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Viral Community Function from Genomic Sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A Novel k-Mer Based Tool for Identifying Viral Sequences from Assembled Metagenomic Data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Pons, J.C.; Paez-Espino, D.; Riera, G.; Ivanova, N.; Kyrpides, N.C.; Llabrés, M. VPF-Class: Taxonomic Assignment and Host Prediction of Uncultivated Viruses Based on Viral Protein Families. Bioinformatics 2021, 37, 1805–1813. [Google Scholar] [CrossRef]
- von Meijenfeldt, F.A.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust Taxonomic Classification of Uncharted Microbial Sequences and Bins with CAT and BAT. Genome Biol. 2019, 20, 217. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Pharokka: A Fast Scalable Bacteriophage Annotation Tool. Bioinformatics 2022, 39, btac776. [Google Scholar] [CrossRef]
- Kurihara, L.; Banks, L.; Chupreta, S.; Couture, C.; Kelchner, V.; Laliberte, J.; Sandhu, S.; Spurbeck, R.; Makarov, V. Abstract 3564: A New Method for Preparation of Low-Input, PCR-Free next Generation Sequencing Libraries. Cancer Res. 2014, 74, 3564. [Google Scholar] [CrossRef]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards Quantitative Viromics for Both Double-Stranded and Single-Stranded DNA Viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef]
- Aigrain, L.; Gu, Y.; Quail, M.A. Quantitation of next Generation Sequencing Library Preparation Protocol Efficiencies Using Droplet Digital PCR Assays—A Systematic Comparison of DNA Library Preparation Kits for Illumina Sequencing. BMC Genom. 2016, 17, 458. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, T.; Zhao, H.; Eterovic, A.K.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. Bias from Removing Read Duplication in Ultra-Deep Sequencing Experiments. Bioinformatics 2014, 30, 1073–1080. [Google Scholar] [CrossRef]
- Cai, L.; Weinbauer, M.G.; Xie, L.; Zhang, R. The Smallest in the Deepest: The Enigmatic Role of Viruses in the Deep Biosphere. Natl. Sci. Rev. 2023, 10, nwad009. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral Metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Dharamshi, J.E.; Tamarit, D.; Eme, L.; Stairs, C.W.; Martijn, J.; Homa, F.; Jørgensen, S.L.; Spang, A.; Ettema, T.J.G. Marine Sediments Illuminate Chlamydiae Diversity and Evolution. Curr. Biol. 2020, 30, 1032–1048.e7. [Google Scholar] [CrossRef]
- Pratama, A.A.; Bolduc, B.; Zayed, A.A.; Zhong, Z.-P.; Guo, J.; Vik, D.R.; Gazitúa, M.C.; Wainaina, J.M.; Roux, S.; Sullivan, M.B. Expanding Standards in Viromics: In Silico Evaluation of dsDNA Viral Genome Identification, Classification, and Auxiliary Metabolic Gene Curation. PeerJ 2021, 9, e11447. [Google Scholar] [CrossRef]
- Danovaro, R.; Dell’Anno, A.; Corinaldesi, C.; Rastelli, E.; Cavicchioli, R.; Krupovic, M.; Noble, R.T.; Nunoura, T.; Prangishvili, D. Virus-Mediated Archaeal Hecatomb in the Deep Seafloor. Sci. Adv. 2016, 2, e1600492. [Google Scholar] [CrossRef]
- Snyder, J.C.; Bolduc, B.; Young, M.J. 40 Years of Archaeal Virology: Expanding Viral Diversity. Virology 2015, 479–480, 369–378. [Google Scholar] [CrossRef]
- Castelán-Sánchez, H.G.; Lopéz-Rosas, I.; García-Suastegui, W.A.; Peralta, R.; Dobson, A.D.W.; Batista-García, R.A.; Dávila-Ramos, S. Extremophile Deep-Sea Viral Communities from Hydrothermal Vents: Structural and Functional Analysis. Mar. Genom. 2019, 46, 16–28. [Google Scholar] [CrossRef]
- Dell’Anno, A.; Corinaldesi, C.; Danovaro, R. Virus Decomposition Provides an Important Contribution to Benthic Deep-Sea Ecosystem Functioning. Proc. Natl. Acad. Sci. USA 2015, 112, E2014–E2019. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J. Virol. 2020, 94, e00582-20. [Google Scholar] [CrossRef]
- Gong, Z.; Liang, Y.; Wang, M.; Jiang, Y.; Yang, Q.; Xia, J.; Zhou, X.; You, S.; Gao, C.; Wang, J.; et al. Viral Diversity and Its Relationship With Environmental Factors at the Surface and Deep Sea of Prydz Bay, Antarctica. Front. Microbiol. 2018, 9, 2981. [Google Scholar] [CrossRef]
- Gazitúa, M.C.; Vik, D.R.; Roux, S.; Gregory, A.C.; Bolduc, B.; Widner, B.; Mulholland, M.R.; Hallam, S.J.; Ulloa, O.; Sullivan, M.B. Potential Virus-Mediated Nitrogen Cycling in Oxygen-Depleted Oceanic Waters. ISME J. 2021, 15, 981–998. [Google Scholar] [CrossRef]
- Chevallereau, A.; Pons, B.J.; van Houte, S.; Westra, E.R. Interactions between Bacterial and Phage Communities in Natural Environments. Nat. Rev. Microbiol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Gao, C.; Liang, Y.; Jiang, Y.; Paez-Espino, D.; Han, M.; Gu, C.; Wang, M.; Yang, Y.; Liu, F.; Yang, Q.; et al. Virioplankton Assemblages from Challenger Deep, the Deepest Place in the Oceans. iScience 2022, 25, 104680. [Google Scholar] [CrossRef]

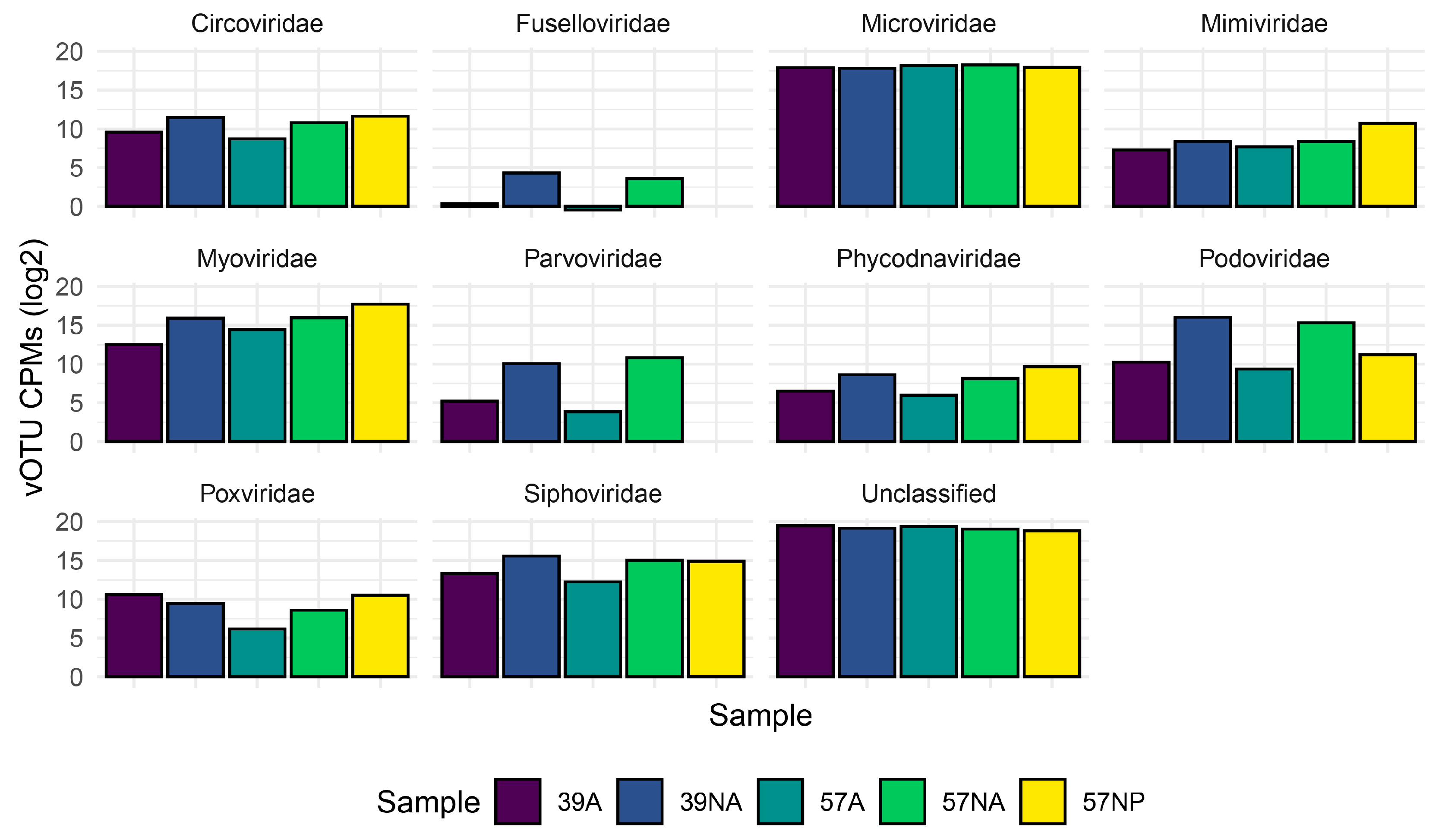
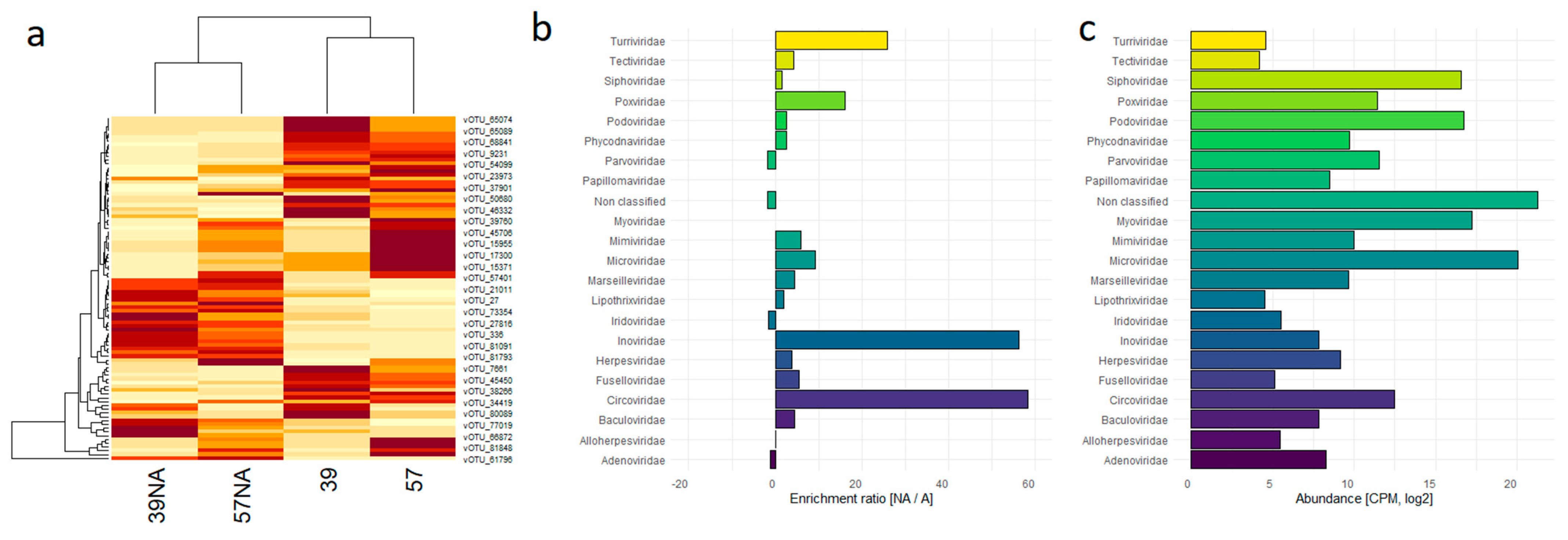

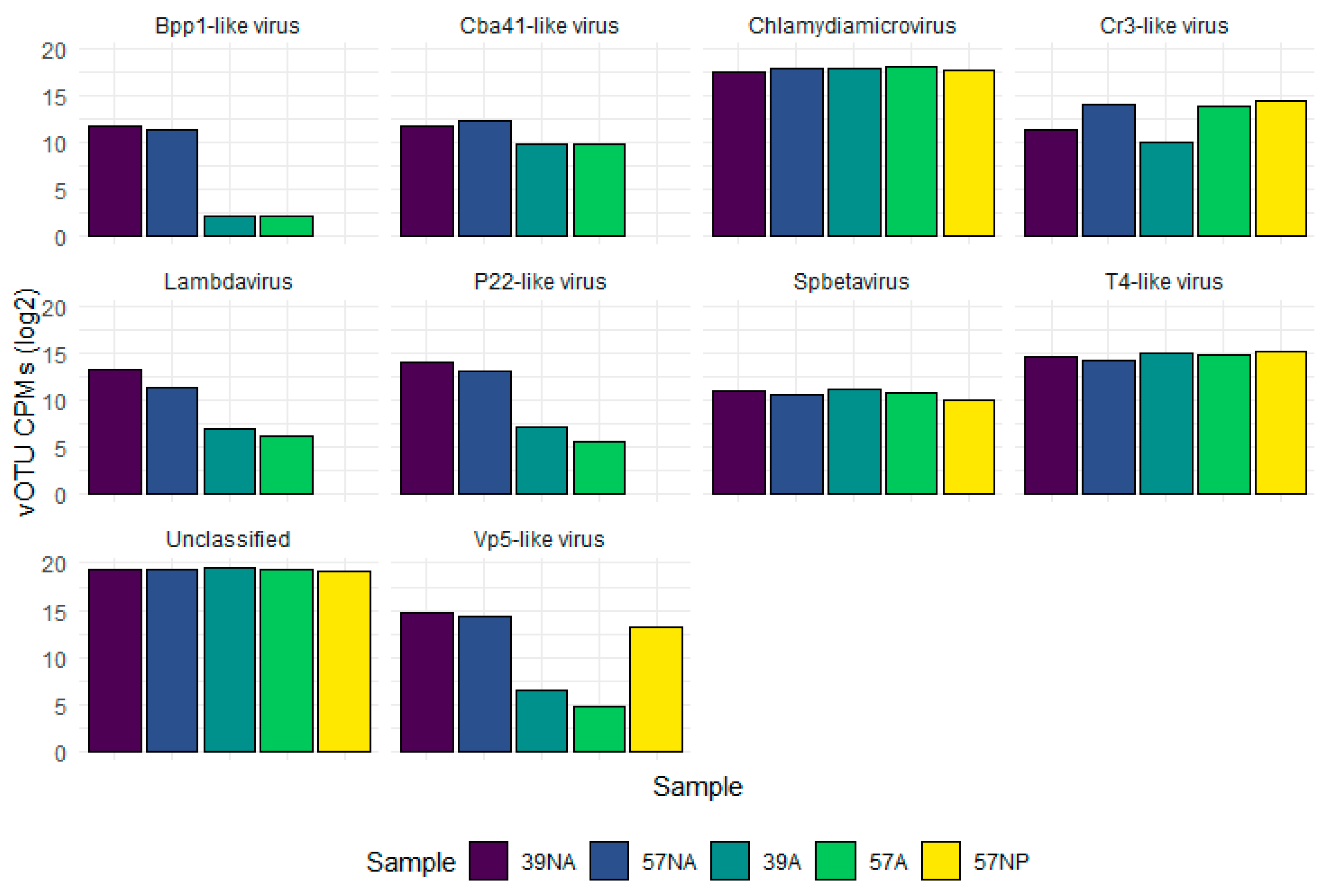

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Description | Technology | Starting DNA Quantity | No. of Deduplicated Reads [×106] | No. of Contigs Longer Than 1.5 kb | Longest Contig (kb) |
|---|---|---|---|---|---|---|
| 39NA | Site 39, not amplified | Illumina | 20–50 pg | 157.3 | 15,199 | 72,980 |
| 39A | Site 39, amplified | Illumina | 10 ng | 488.0 | 79,174 | 151,451 |
| 57NA | Site 57, not amplified | Illumina | 20–50 pg | 211.0 | 35,891 | 135,810 |
| 57A | Site 57, amplified | Illumina | 10 ng | 471.0 | 62,878 | 135,810 |
| Nanopore | 1 µg | 0.331 | 1009 |
| Sample ID | No. of vOTUs | Total Reads Mapped Back on vOTUs | Richness Chao1 | Alpha Diversity Shannon | Alpha Diversity Fisher |
|---|---|---|---|---|---|
| 39NA | 11,031 | 4.4% | 10,626 | 7.11 | 1661.12 |
| 39A | 10,393 | 38.5% | 9662 | 6.96 | 1487.58 |
| 57NA | 12,354 | 5.8% | 11,760 | 7.30 | 1875.05 |
| 57A | 10,346 | 49.9% | 9659 | 6.86 | 1486.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosani, U.; Corinaldesi, C.; Luongo, G.; Sollitto, M.; Dal Monego, S.; Licastro, D.; Bongiorni, L.; Venier, P.; Pallavicini, A.; Dell’Anno, A. Viral Diversity in Benthic Abyssal Ecosystems: Ecological and Methodological Considerations. Viruses 2023, 15, 2282. https://doi.org/10.3390/v15122282
Rosani U, Corinaldesi C, Luongo G, Sollitto M, Dal Monego S, Licastro D, Bongiorni L, Venier P, Pallavicini A, Dell’Anno A. Viral Diversity in Benthic Abyssal Ecosystems: Ecological and Methodological Considerations. Viruses. 2023; 15(12):2282. https://doi.org/10.3390/v15122282
Chicago/Turabian StyleRosani, Umberto, Cinzia Corinaldesi, Gabriella Luongo, Marco Sollitto, Simeone Dal Monego, Danilo Licastro, Lucia Bongiorni, Paola Venier, Alberto Pallavicini, and Antonio Dell’Anno. 2023. "Viral Diversity in Benthic Abyssal Ecosystems: Ecological and Methodological Considerations" Viruses 15, no. 12: 2282. https://doi.org/10.3390/v15122282