
Recent Advances in the Development of Sulfamoyl-Based Hepatitis B Virus Nucleocapsid Assembly Modulators

 , , and
, , and
Abstract
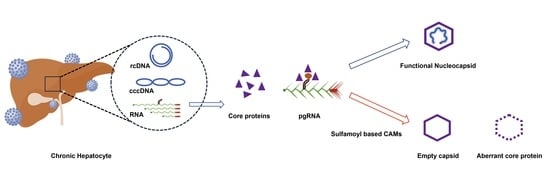
:
1. Introduction
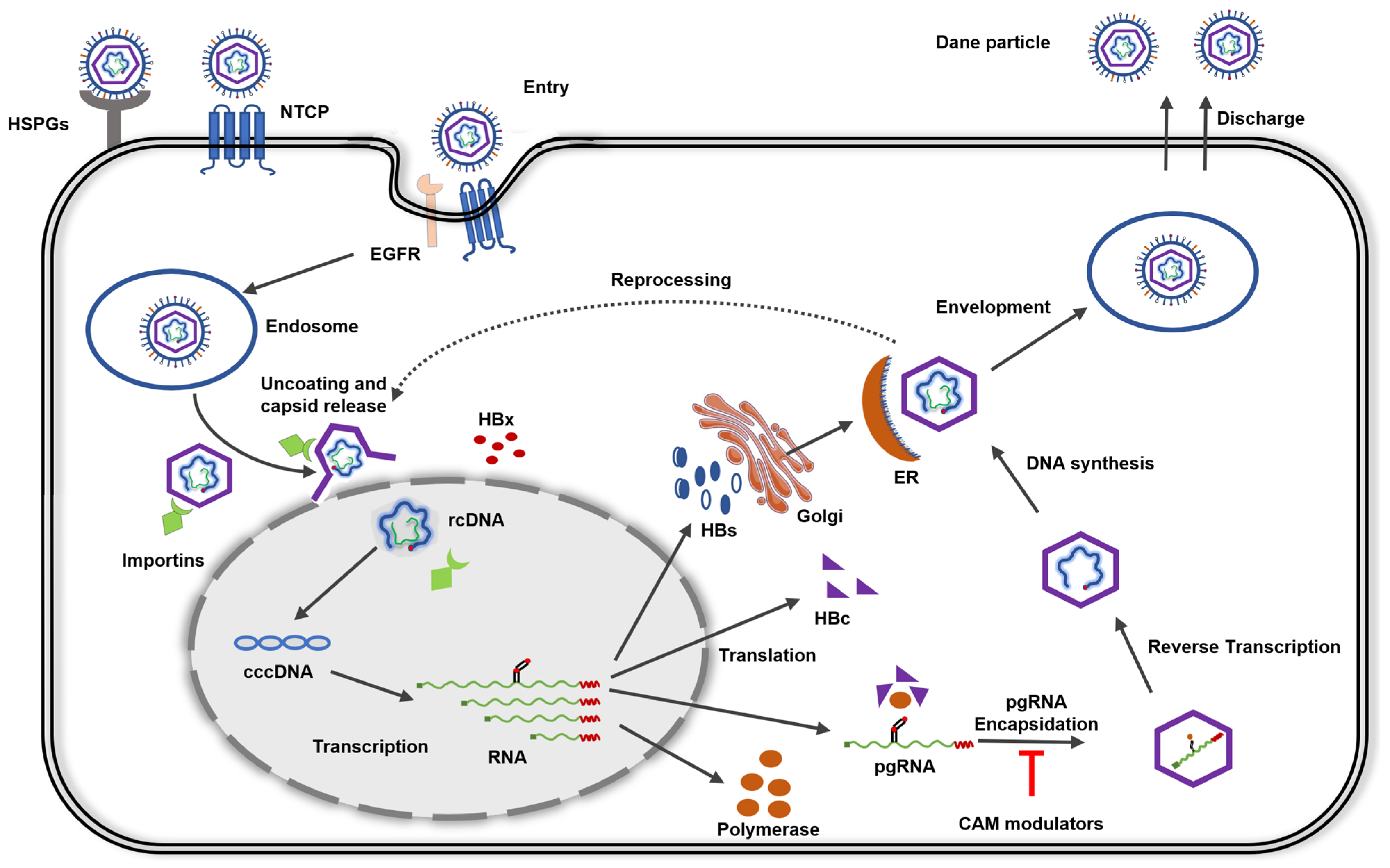
2. HBV Life Cycle
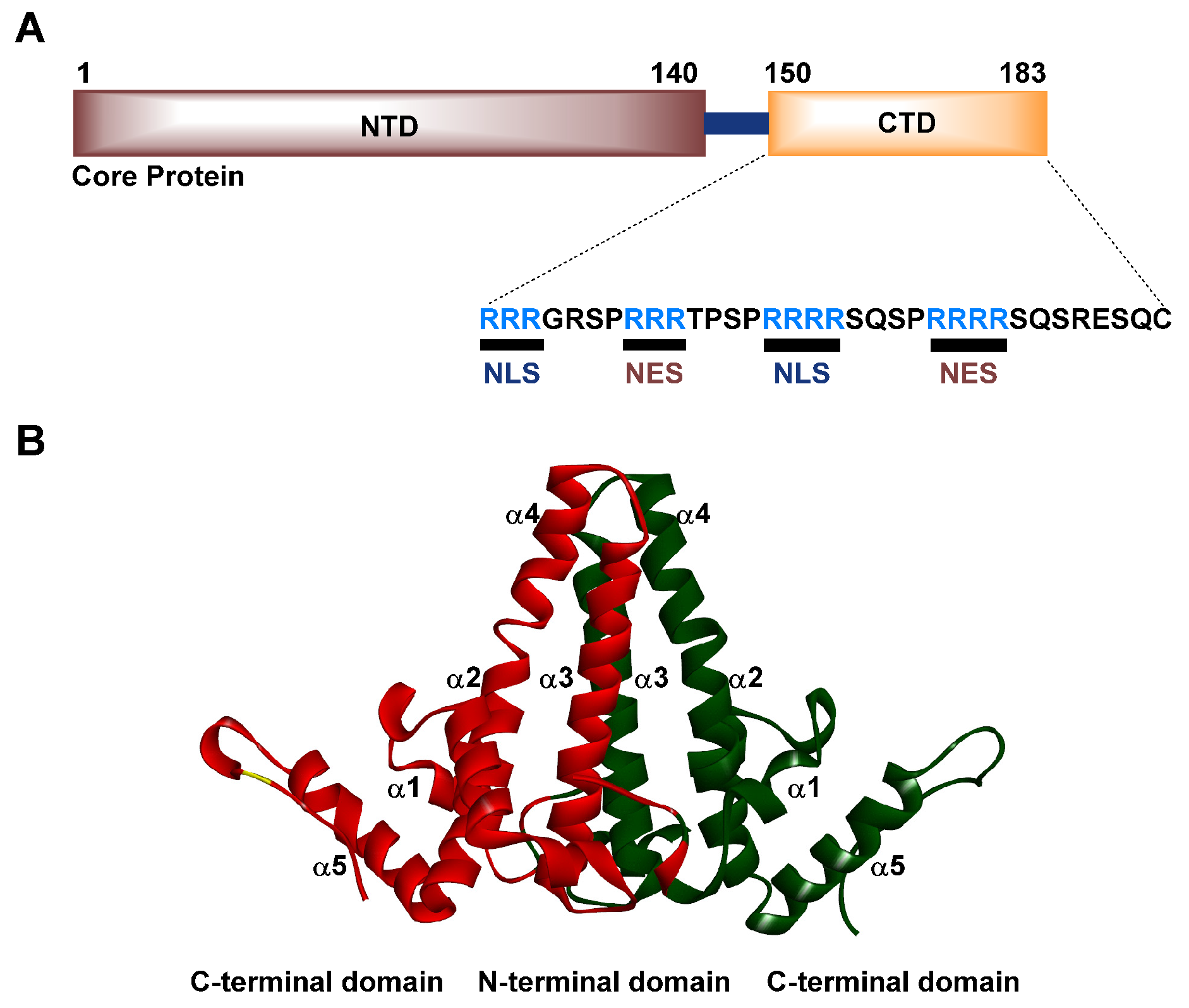
3. Structure and Function of the HBV Cp
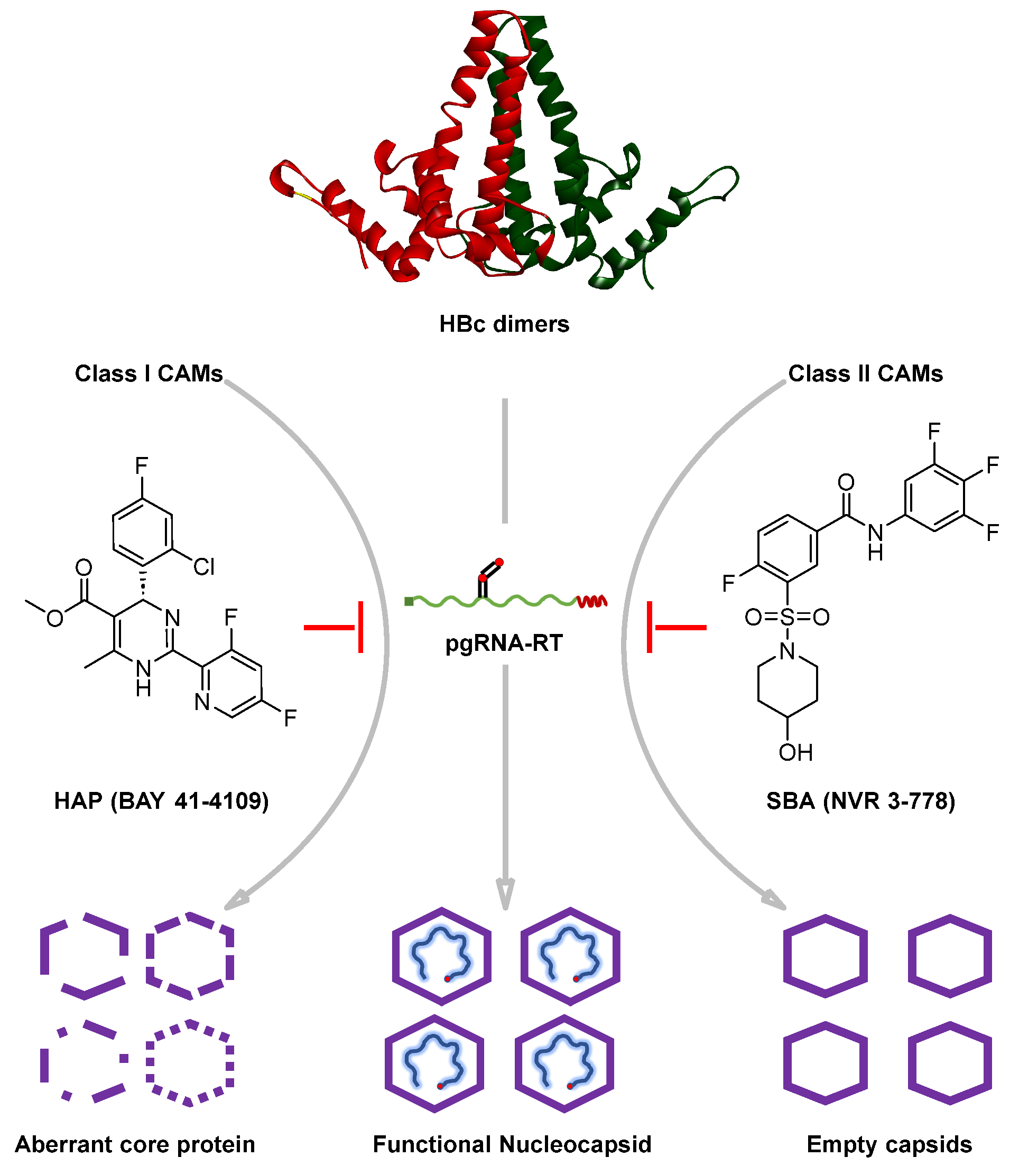
4. Influence of CAMs on Capsid Assembly: Classification of CAMs
4.1. Frequently Used In Vitro Cell Culture Models for HBV Infection Studies
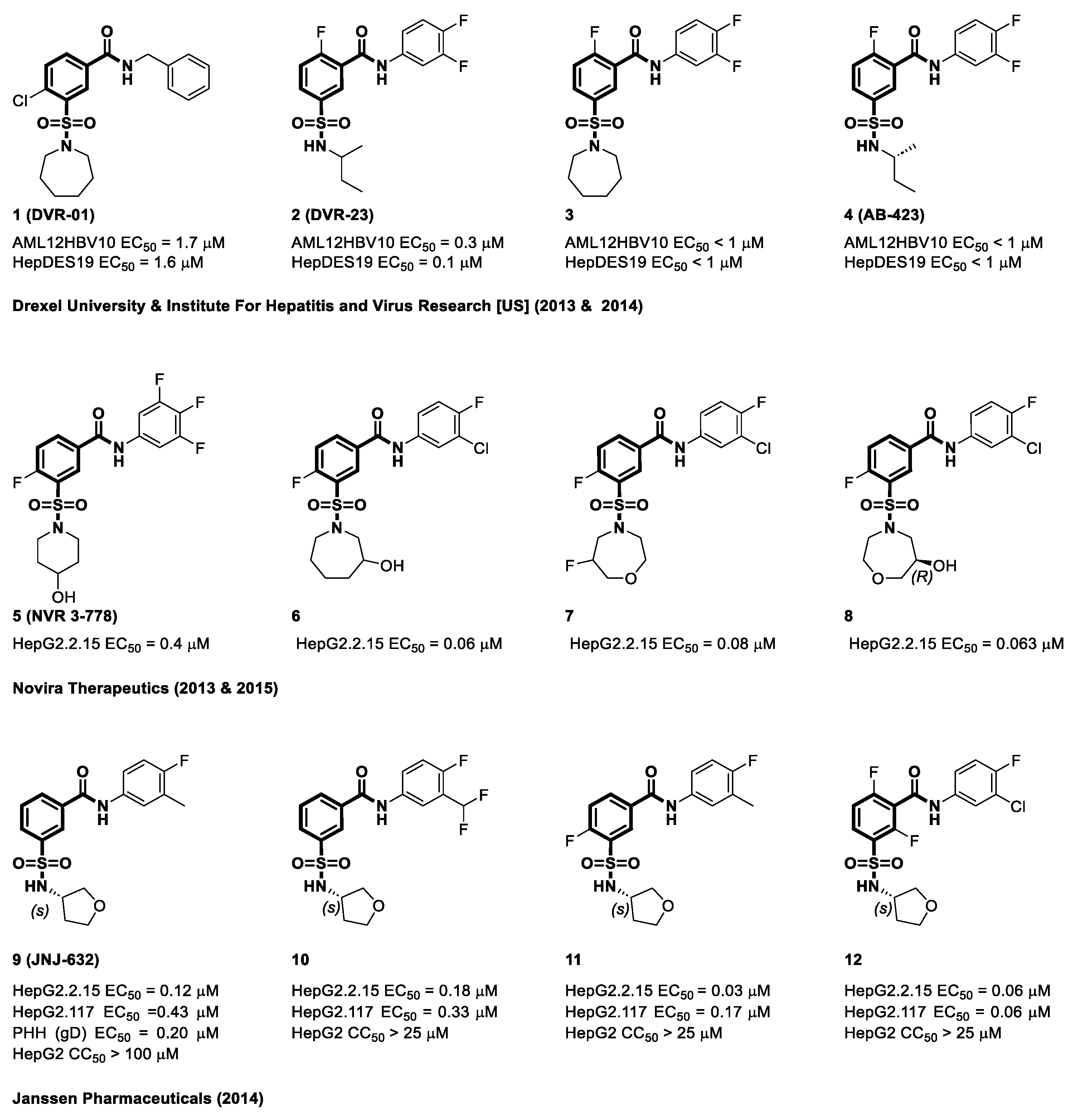
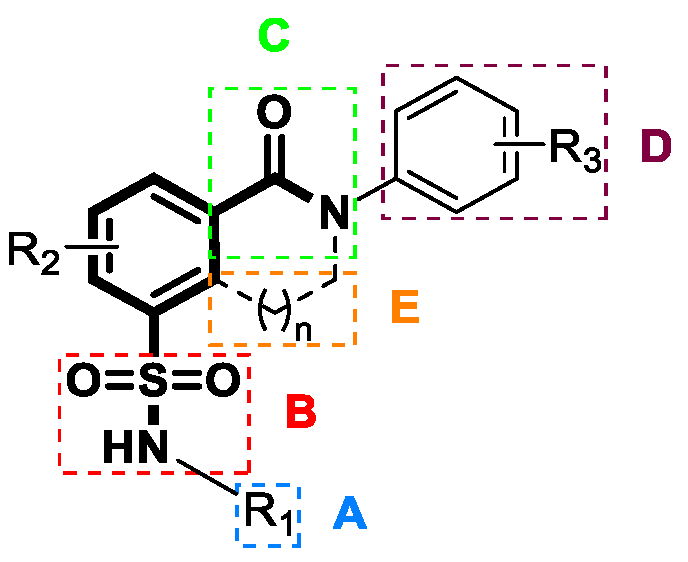
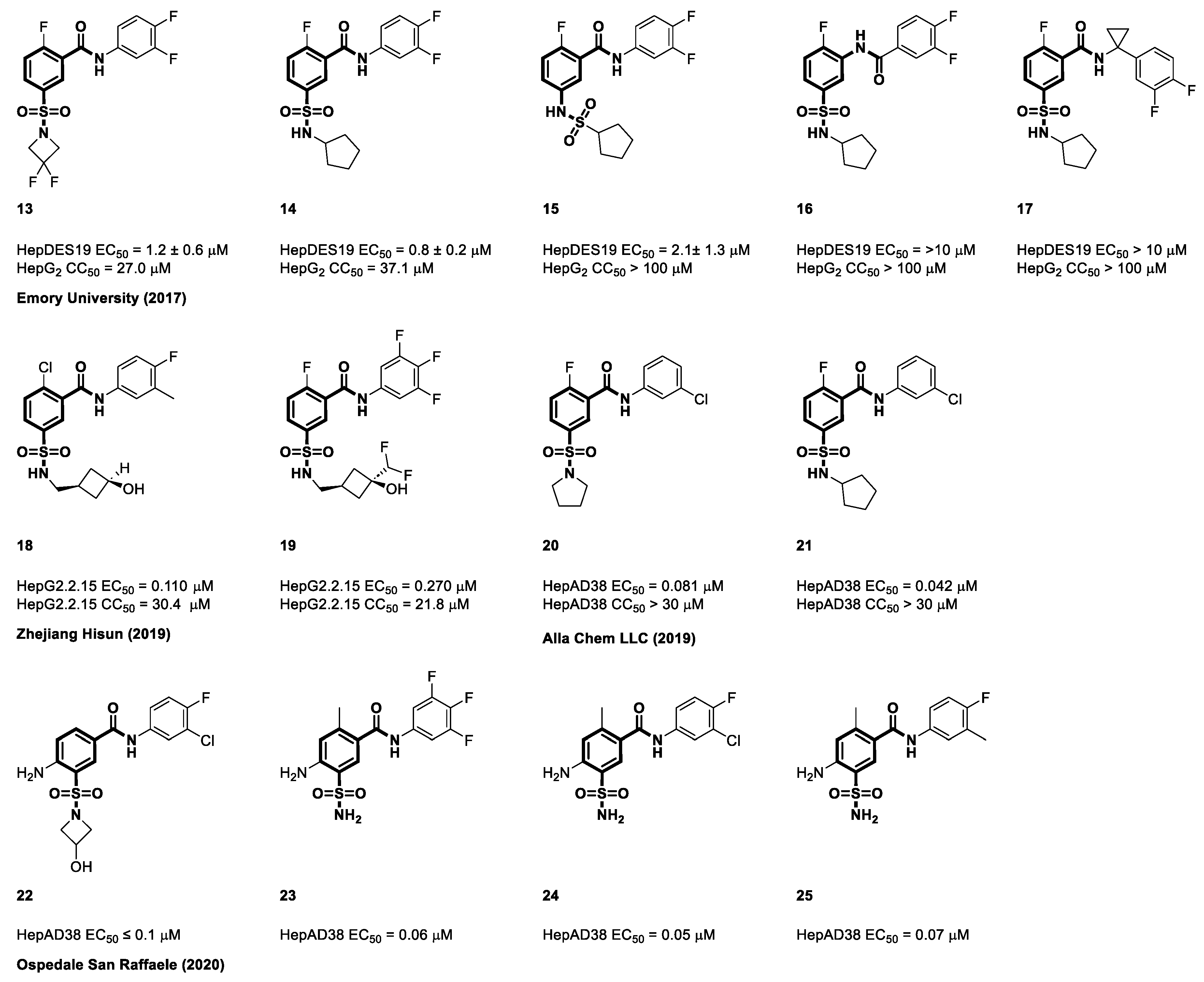
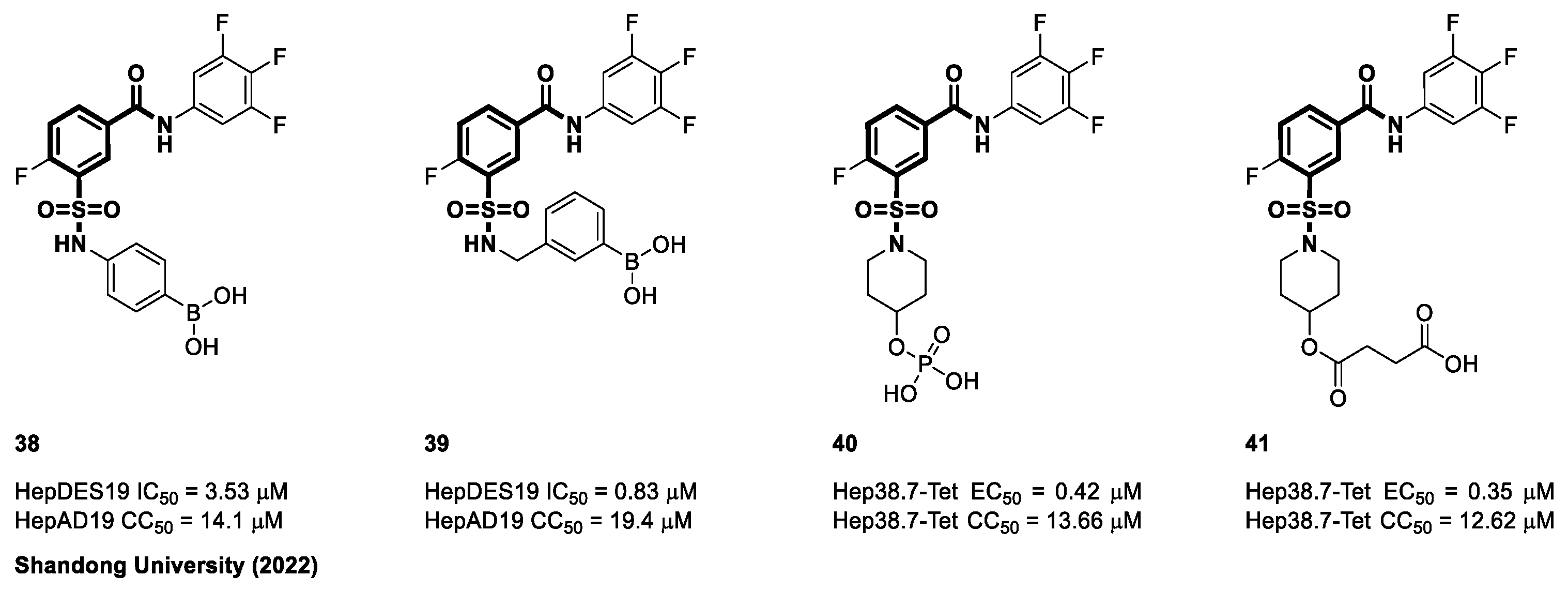
4.2. Sulfamoyl Benzocarboxamides as CAMs
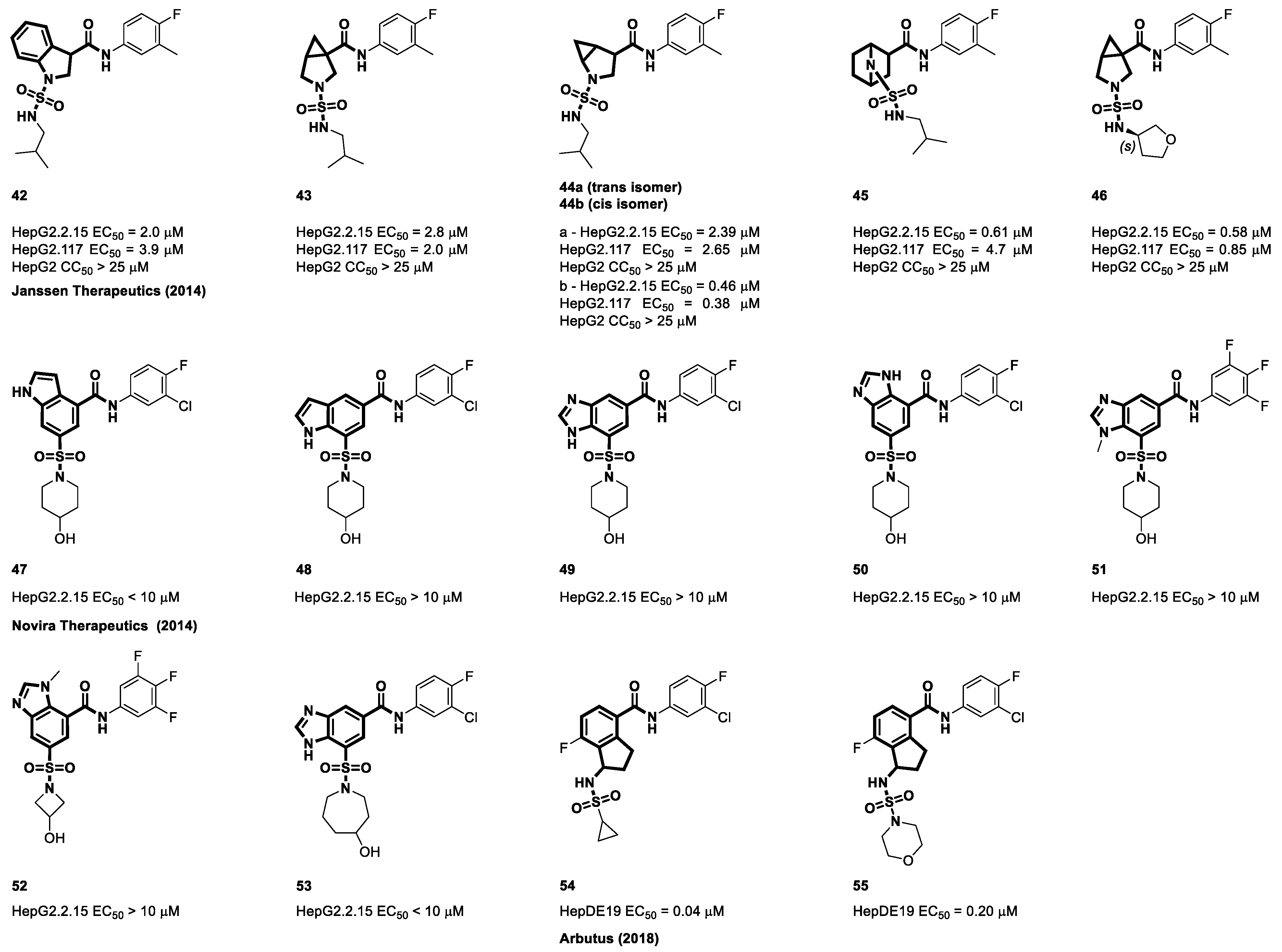
4.3. Sulfamoyl Bicyclic Carboxamide-Based CAMs
4.4. Sulfamoyl Aliphatic Heterocyclic-Carboxamide-Based CAMs
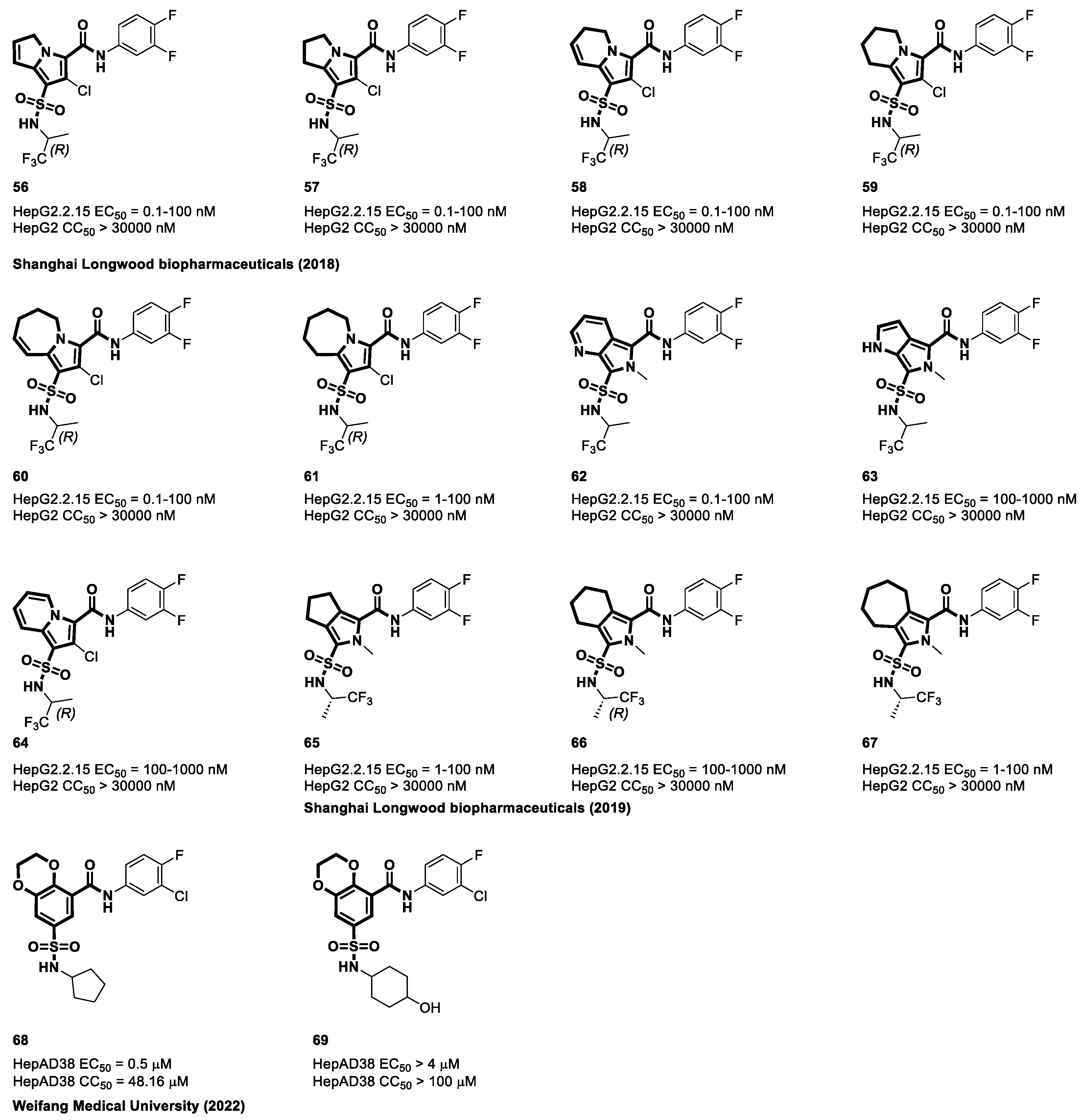
4.5. Sulfamoyl Aromatic Heterocyclic-Carboxamide-Based CAMs
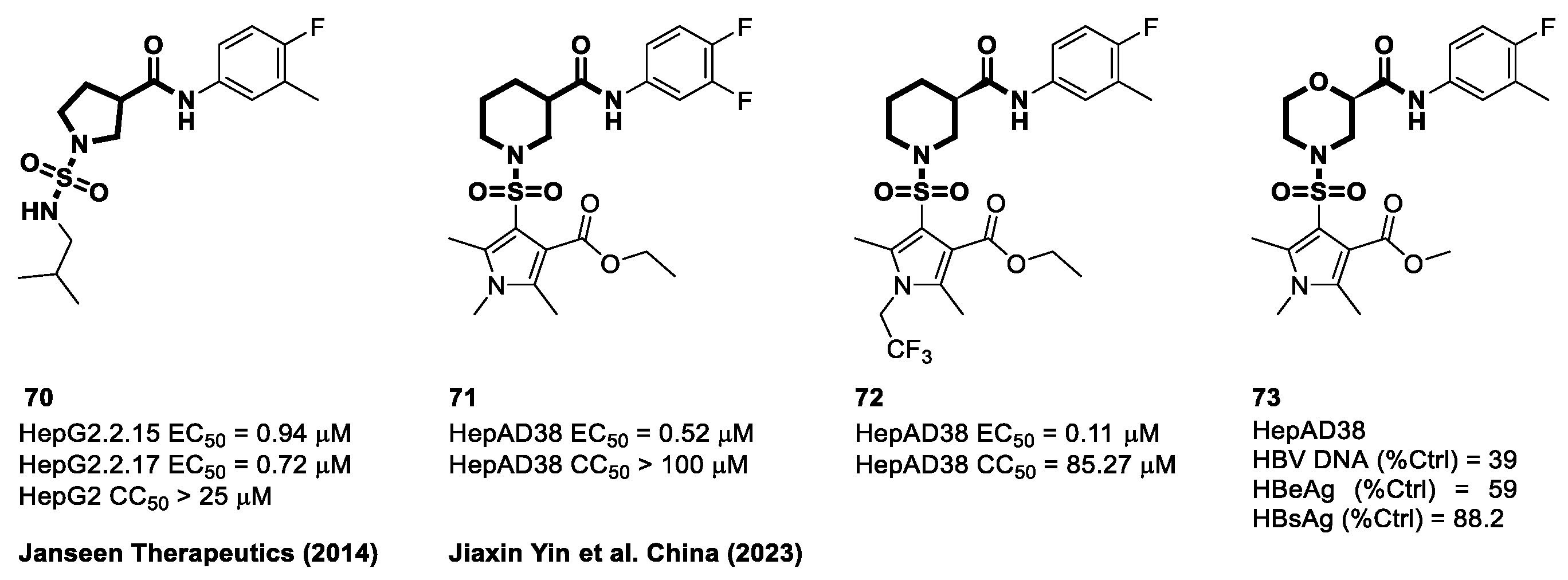
4.6. Cyclic Sulfonamide Carboxamide-Based CAMs
4.7. Non-Carboxamide Sulfomoyl-Based CAMs
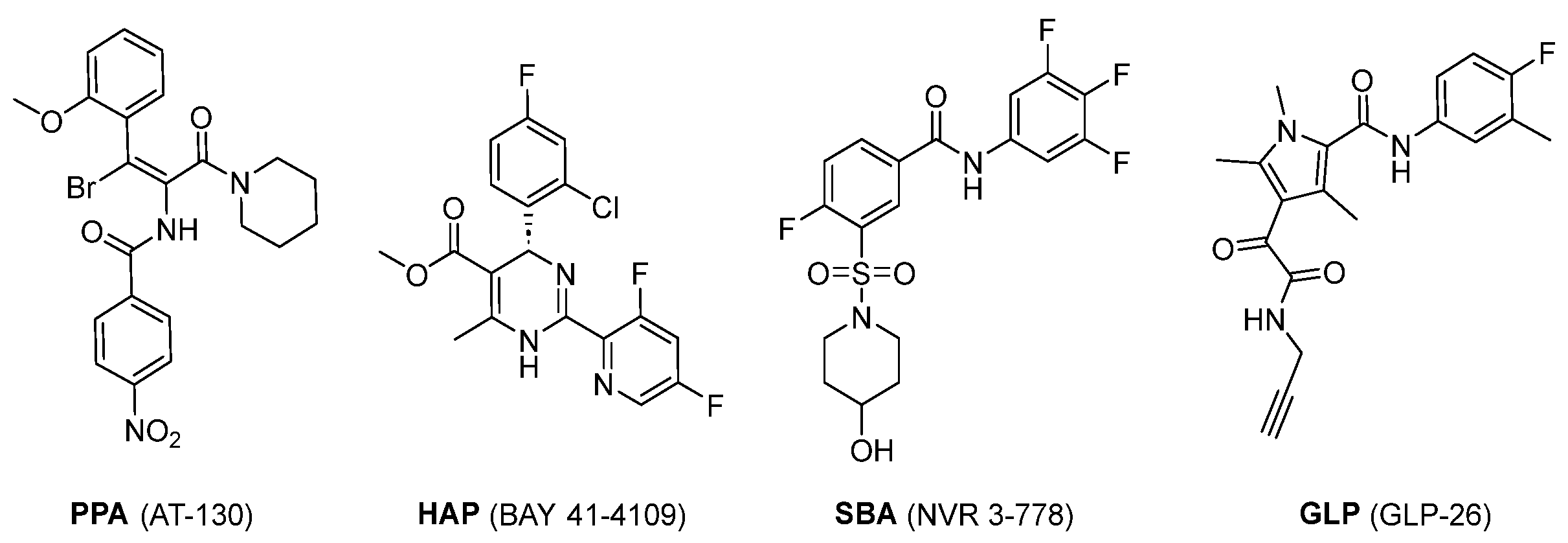
5. Sulfamoyl-Based Carboxamide CAMs in Clinical Development
5.1. NVR 3-778
5.2. JNJ56136379
5.3. ABI-H0731
5.4. AB-423
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fauquet, C.M.; Fargette, D. International Committee on Taxonomy of Viruses and the 3142 Unassigned Species. Virol. J. 2005, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S. Hepatitis B Virus Taxonomy and Hepatitis B Virus Genotypes. World J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B Virus Molecular Biology and Pathogenesis. Hepatoma Res. 2016, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Guidotti, L.G. Immunobiology and Pathogenesis of Hepatitis B Virus Infection. Nat. Rev. Immunol. 2022, 22, 19–32. [Google Scholar] [CrossRef]
- Stasi, C.; Silvestri, C.; Voller, F. Hepatitis B Vaccination and Immunotherapies: An Update. Clin. Exp. Vaccine Res. 2020, 9, 1–7. [Google Scholar] [CrossRef]
- Guvenir, M.; Arikan, A. Hepatitis B Virus: From Diagnosis to Treatment. Polish J. Microbiol. 2020, 69, 391–399. [Google Scholar] [CrossRef]
- Thibault, V.; Laperche, S.; Thiers, V.; Sayon, S.; Letort, M.J.; Delarocque-Astagneau, E.; Antona, D. Molecular Epidemiology and Clinical Characteristics of Hepatitis B Identified through the French Mandatory Notification System. PLoS ONE 2013, 8, e75267. [Google Scholar] [CrossRef]
- Lemon, S.M. What Is the Role of Testing for IgM Antibody to Core Antigen of Hepatitis B Virus? Mayo Clin. Proc. 1988, 63, 201–204. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular Biology of Hepatitis B Virus Infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of Viral Hepatitis and Hepatocellular Carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef]
- Woo, A.S.J.; Kwok, R.; Ahmed, T. Alpha-Interferon Treatment in Hepatitis B. Ann. Transl. Med. 2017, 5, 159. [Google Scholar] [CrossRef]
- Kim, S.S.; Cheong, J.Y.; Cho, S.W. Current Nucleos(t)ide Analogue Therapy for Chronic Hepatitis B. Gut Liver 2011, 5, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.; Lai, C.L.; Seto, W.K.; Yuen, M.F. Nucleoside/Nucleotide Analogues in the Treatment of Chronic Hepatitis B. J. Antimicrob. Chemother. 2011, 66, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.T.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 Cleavage of Viral DNA Efficiently Suppresses Hepatitis B Virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Bonnin, M.; Aillot, L.; Fusil, F.; Maadadi, S.; Dimier, L.; Michelet, M.; Floriot, O.; Ollivier, A.; Rivoire, M.; et al. Direct Antiviral Properties of TLR Ligands against HBV Replication in Immune-Competent Hepatocytes. Sci. Rep. 2018, 8, 5390. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Ren, S.; Espiritu, C.; Kelly, M.; Lau, V.; Zheng, L.; Hartman, G.D.; Flores, O.A. Hepatitis B Virus Capsid Assembly Modulators, but Not Nucleoside Analogs, Inhibit the Production of Extracellular Pregenomic RNA and Spliced RNA Variants. Antimicrob. Agents Chemother. 2017, 61, e00680-17. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Suzuki, T.; Ishibashi, R.; Seimiya, T.; Tanaka, E.; Koike, K. Inhibition of HBV Transcription From cccDNA with Nitazoxanide by Targeting the HBx–DDB1 Interaction. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 297–312. [Google Scholar] [CrossRef]
- Su, L. Disruption of HBx-DDB1 by NTZ: New Mechanistic Insight into an Old Drug With Broad Anti-Infective Activities. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 289–290. [Google Scholar] [CrossRef]
- Chang, J.; Guo, F.; Zhao, X.; Guo, J.-T. Therapeutic Strategies for a Functional Cure of Chronic Hepatitis B Virus Infection. Acta Pharm. Sin. B 2014, 4, 248–257. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core Protein: A Pleiotropic Keystone in the HBV Lifecycle. Antivir. Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Deres, K.; Schroder, C.H.; Paessens, A.; Goldmann, S.; Hacker, J.H.; Weber, O.; Kramer, T.; Niewohner, U.; Pleiss, U.; Stoltefuss, J.; et al. Inhibition of Hepatitis B Virus Replication by Drug-Induced Depletion of Nucleocapsids. Science 2003, 299, 893–896. [Google Scholar] [CrossRef]
- Perni, R.B.; Conway, S.C.; Ladner, S.K.; Zaifert, K.; Otto, M.J.; King, R.W. Phenylpropenamide Derivatives as Inhibitors of Hepatitis B Virus Replication. Bioorg. Med. Chem. Lett. 2000, 10, 2687–2690. [Google Scholar] [CrossRef] [PubMed]
- King, R.W.; Ladner, S.K.; Miller, T.J.; Zaifert, K.; Perni, R.B.; Conway, S.C.; Otto, M.J. Erratum: Inhibition of Human Hepatitis B Virus Replication by AT-61, a Phenylpropenamide Derivative, Alone and in Combination with (-)β-L-2′,3′- Dideoxy-3′-Thiacytidine. Antimicrob. Agents Chemother. 1998, 42, 3179–3186. [Google Scholar] [CrossRef] [PubMed]
- Weber, O.; Schlemmer, K.H.; Hartmann, E.; Hagelschuer, I.; Paessens, A.; Graef, E.; Deres, K.; Goldmann, S.; Niewoehner, U.; Stoltefuss, J.; et al. Inhibition of Human Hepatitis B Virus (HBV) by a Novel Non-Nucleosidic Compound in a Transgenic Mouse Model. Antivir. Res. 2002, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Amblard, F.; Boucle, S.; Bassit, L.; Chen, Z.; Sari, O.; Cox, B.; Verma, K.; Ozturk, T.; Ollinger-Russell, O.; Schinazi, R.F. Discovery and Structure Activity Relationship of Glyoxamide Derivatives as Anti-Hepatitis B Virus Agents. Bioorg. Med. Chem. 2021, 31, 115952. [Google Scholar] [CrossRef]
- Campagna, M.R.; Liu, F.; Mao, R.; Mills, C.; Cai, D.; Guo, F.; Zhao, X.; Ye, H.; Cuconati, A.; Guo, H.; et al. Sulfamoylbenzamide Derivatives Inhibit the Assembly of Hepatitis B Virus Nucleocapsids. J. Virol. 2013, 87, 6931–6942. [Google Scholar] [CrossRef]
- Nagraj, M.; Andrrew, G.C.; Janet, R.P.; Andrzej, A.; Dorsey, B.D.; Evangelista, E.; Fan, K.; Guo, F.; Guo, H.; Guo, J.; et al. Preclinical Profile of AB-423, an Inhibitor of Hepatitis B Virus Pregenomic RNA Encapsidation. Agents Chemother. 2018, 62, e00082-18. [Google Scholar] [CrossRef]
- Lam, A.M.; Espiritu, C.; Vogel, R.; Ren, S.; Lau, V.; Kelly, M.; Kuduk, S.D.; Hartman, G.D.; Flores, O.A.; Klumpp, K. Preclinical Characterization of NVR 3-778, a First-in-Class Capsid Assembly Modulator against Hepatitis B Virus. Antimicrob. Agents Chemother. 2019, 63, e01734-18. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K. Hepatitis B Virus Biology and Life Cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of Glycosaminoglycans for Binding and Infection of Hepatitis B Virus. Cell. Microbiol. 2008, 10, 122–133. [Google Scholar] [CrossRef]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B Virus Infection Initiates with a Large Surface Protein-Dependent Binding to Heparan Sulfate Proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoudé, G.; et al. A Targeted Functional RNA Interference Screen Uncovers Glypican 5 as an Entry Factor for Hepatitis B and D Viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium Taurocholate Cotransporting Polypeptide is a Functional Receptor for Human Hepatitis B and D Virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal Growth Factor Receptor Is a Host-Entry Cofactor Triggering Hepatitis B Virus Internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.-H.; Park, S.-Y.; Ohshima, T.; et al. The Machinery for Endocytosis of Epidermal Growth Factor Receptor Coordinates the Transport of Incoming Hepatitis B Virus to the Endosomal Network. J. Biol. Chem. 2020, 295, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Fukano, K.; Tsukuda, S.; Oshima, M.; Suzuki, R.; Aizaki, H.; Ohki, M.; Park, S.Y.; Muramatsu, M.; Wakita, T.; Sureau, C.; et al. Troglitazone Impedes the Oligomerization of Sodium Taurocholate Cotransporting Polypeptide and Entry of Hepatitis B Virus into Hepatocytes. Front. Microbiol. 2019, 10, 3257. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.; Glebe, D.; Kann, M. Lipid-Mediated Introduction of Hepatitis B Virus Capsids into Nonsusceptible Cells Allows Highly Efficient Replication and Facilitates the Study of Early Infection Events. J. Virol. 2006, 80, 5465–5473. [Google Scholar] [CrossRef]
- Kann, M.; Sodeik, B.; Vlachou, A.; Gerlich, W.H.; Helenius, A. Phosphorylation-Dependent Binding of Hepatitis B Virus Core Particles to the Nuclear Pore Complex. J. Cell Biol. 1999, 145, 45–55. [Google Scholar] [CrossRef]
- Wei, L.; Ploss, A. Hepatitis B Virus cccDNA is Formed through Distinct Repair Processes of Each Strand. Nat. Commun. 2021, 12, 1591–1603. [Google Scholar] [CrossRef]
- Sheraz, M.; Cheng, J.; Tang, L.; Chang, J.; Guo, J.T. Cellular DNA Topoisomerases are Required for the Synthesis of Hepatitis B Virus Covalently Closed Circular DNA. J. Virol. 2019, 93, e02230-18. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap Endonuclease 1 is Involved in cccDNA Formation in the Hepatitis B Virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef] [PubMed]
- KöNiger, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the Host DNA-Repair Enzyme TDP2 in Formation of the Covalently Closed Circular DNA Persistence Reservoir of Hepatitis B Viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Kim, E.S.; Guo, H. Epigenetic Regulation of Hepatitis B Virus Covalently Closed Circular DNA: Implications for Epigenetic Therapy against Chronic Hepatitis B. Hepatology 2017, 66, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Seeger, C. Novel Mechanism for Reverse Transcription in Hepatitis B Viruses. J. Virol. 1993, 67, 6507–6512. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Schaller, H. Hepadnaviral Assembly is Initiated by Polymerase Binding to the Encapsidation Signal in the Viral RNA Genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Junker-Niepmann, M.; Schaller, H. The P Gene Product of Hepatitis B Virus is Required as a Structural Component for Genomic RNA Encapsidation. J. Virol. 1990, 64, 5324–5332. [Google Scholar] [CrossRef]
- Rall, L.B.; Standring, D.N.; Laub, O.; Rutter, W.J. Transcription of Hepatitis B Virus by RNA Polymerase II. Mol. Cell. Biol. 1983, 3, 1766–1773. [Google Scholar] [CrossRef]
- Beck, J.; Nassal, M. Hepatitis B Virus Replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef]
- Hu, J. Hepatitis B Virus Virology and Replication; Humana Press: Cham, Switzerland, 2016; ISBN 9783319223308. [Google Scholar]
- Nassal, M.; Schaller, H. Hepatitis B Virus Replication. Trends Microbiol. 1993, 1, 221–228. [Google Scholar] [CrossRef]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G.W. The Crystal Structure of the Human Hepatitis B Virus Capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Seeger, C. Hepadnavirus Genome Replication and Persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral Persistence Reservoir and Key Obstacle for a Cure of Chronic Hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. Hepatitis B Viruses: Reverse Transcription a Different Way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of Host Cellular Multivesicular Body Functions in Hepatitis B Virus Budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B Virus Genome Recycling and de Novo Secondary Infection Events Maintain Stable cccDNA Levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- de Rocquigny, H.; Rat, V.; Pastor, F.; Darlix, J.L.; Hourioux, C.; Roingeard, P. Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid. Viruses 2020, 12, 738. [Google Scholar] [CrossRef]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear Export and Import of Human Hepatitis B Virus Capsid Protein and Particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef]
- Rat, V.; Pinson, X.; Seigneuret, F.; Durand, S.; Herrscher, C.; Lemoine, R.; Burlaud-Gaillard, J.; Raynal, P.Y.; Hourioux, C.; Roingeard, P.; et al. Hepatitis B Virus Core Protein Domains Essential for Viral Capsid Assembly in a Cellular Context. J. Mol. Biol. 2020, 432, 3802–3819. [Google Scholar] [CrossRef]
- Ludgate, L.; Liu, K.; Luckenbaugh, L.; Streck, N.; Eng, S.; Voitenleitner, C.; Delaney, W.E.; Hu, J. Cell-Free Hepatitis B Virus Capsid Assembly Dependent on the Core Protein C-Terminal Domain and Regulated by Phosphorylation. J. Virol. 2016, 90, 5830–5844. [Google Scholar] [CrossRef]
- Liu, K.; Luckenbaugh, L.; Ning, X.; Xi, J.; Hu, J. Multiple Roles of Core Protein Linker in Hepatitis B Virus Replication. PLoS Pathog. 2018, 14, e1007085. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci. Rep. 2017, 7, 42374. [Google Scholar] [CrossRef]
- Stray, S.J.; Zlotnick, A. BAY 41-4109 has Multiple Effects on Hepatitis B Virus Capsid Assembly. J. Mol. Recognit. 2006, 19, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, T.; Huang, X.; Kumar, G.R.; Chen, X.; Zeng, Z.; Zhang, R.; Chen, R.; Li, T.; Zhang, T.; et al. Serum Hepatitis B Virus RNA is Encapsidated Pregenome RNA that may be Associated with Persistence of Viral Infection and Rebound. J. Hepatol. 2016, 65, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Mukhopadhyay, S. Virus Assembly, Allostery and Antivirals. Trends Microbiol. 2011, 19, 14–23. [Google Scholar] [CrossRef]
- Tan, Z.; Maguire, M.L.; Loeb, D.D.; Zlotnick, A. Genetically Altering the Thermodynamics and Kinetics of Hepatitis B Virus Capsid Assembly has Profound Effects on Virus Replication in Cell Culture. J. Virol. 2013, 87, 3208–3216. [Google Scholar] [CrossRef]
- Bourne, C.R.; Finn, M.G.; Zlotnick, A. Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. J. Virol. 2006, 80, 11055–11061. [Google Scholar] [CrossRef]
- Stray, S.J.; Bourne, C.R.; Punna, S.; Lewis, W.G.; Finn, M.G.; Zlotnick, A. A Heteroaryldihydropyrimidine Activates and can Misdirect Hepatitis B Virus Capsid Assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 8138–8143. [Google Scholar] [CrossRef]
- Venkatakrishnan, B.; Katen, S.P.; Francis, S.; Chirapu, S.; Finn, M.G.; Zlotnick, A. Hepatitis B Virus Capsids have Diverse Structural Responses to Small-Molecule Ligands Bound to the Heteroaryldihydropyrimidine Pocket. J. Virol. 2016, 90, 3994–4004. [Google Scholar] [CrossRef]
- Sarah, P.K.; Reddy, S.C.; Finn, M.G.; Zlotnick, A. Trapping of Hepatitis B Virus Capsid Assembly Article Intermediates by Phenylpropenamide Assembly Accelerators. ACS Chem. Biol. 2010, 5, 1125–1136. [Google Scholar] [CrossRef]
- Bourne, C.; Lee, S.; Venkataiah, B.; Lee, A.; Korba, B.; Finn, M.G.; Zlotnick, A. Small-Molecule Effectors of Hepatitis B Virus Capsid Assembly Give Insight into Virus Life Cycle. J. Virol. 2008, 82, 10262–10270. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Pionek, K.; Unchwaniwala, N.; Maguire, M.L.; Loeb, D.D.; Zlotnick, A. The Interface between Hepatitis B Virus Capsid Proteins Affects Self-Assembly, Pregenomic RNA Packaging, and Reverse Transcription. J. Virol. 2015, 89, 3275–3284. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Hadden, J.A.; Zlotnick, A. Assembly Properties of Hepatitis B Virus Core Protein Mutants Correlate with their Resistance to Assembly-Directed Antivirals. J. Virol. 2018, 92, e01082-18. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhao, Q.; Sheraz, M.; Cheng, J.; Qi, Y.; Su, Q.; Cuconati, A.; Wei, L.; Du, Y.; Li, W.; et al. HBV Core Protein Allosteric Modulators Differentially Alter cccDNA Biosynthesis from de Novo Infection and Intracellular Amplification Pathways. PLoS Pathog. 2017, 13, e1006658. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yan, L.; Shi, Y.; Lv, D.; Shang, J.; Bai, L.; Tang, H. Hepatitis Infection B Virus: Molecular Virology to Antiviral Drugs; Springer: Singapore, 2020; Volume 1179, ISBN 9789811391507. [Google Scholar]
- Zhang, Y.; Mao, R.; Yan, R.; Cai, D.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y. Transcription of Hepatitis B Virus Covalently Closed Circular DNA is Regulated by CpG Methylation during Chronic Infection. PLoS ONE 2014, 9, e110442. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.C.; Bagnaninchi, P.; Ye, H.; Sancho-Bru, P.; Falcon-Perez, J.M.; Royo, F.; Garcia-Ruiz, C.; Konu, O.; Miranda, J.; Lunov, O. Advanced Preclinical Models for Evaluation of Drug-Induced Liver Injury—Consensus Statement by the European Drug-Induced Liver Injury Network. J. Hepatol. 2021, 75, 935–959. [Google Scholar] [CrossRef] [PubMed]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of Hepatitis B Virus Particles in Hep G2 Cells Transfected with Cloned Hepatitis B Virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.Y.; Liu, F.; Seto, W.K.; Lai, C.L.; Yuen, M.F.; Wong, D.K.H. Role of Hepatitis B Core Protein in HBV Transcription and Recruitment of Histone Acetyltransferases to cccDNA Minichromosome. Antiviral Res. 2017, 144, 1–7. [Google Scholar] [CrossRef]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M. Early Inhibition of Hepatocyte Innate Responses by Hepatitis B Virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, L.; Liu, W.; Ma, X.; Cen, J.; Sun, Z.; Wang, C.; Feng, S.; Zhang, Z.; Yue, L. In Vitro Expansion of Primary Human Hepatocytes with Efficient Liver Repopulation Capacity. Cell Stem Cell 2018, 23, 806–819. [Google Scholar] [CrossRef]
- Touboul, T.; Chen, S.; To, C.C.; Mora-Castilla, S.; Sabatini, K.; Tukey, R.H.; Laurent, L.C. Stage-Specific Regulation of the WNT/β-Catenin Pathway Enhances Differentiation of HESCs into Hepatocytes. J. Hepatol. 2016, 64, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schoneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J. HBV Bypasses the Innate Immune Response and does not Protect HCV from Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Filzmayer, C.; Ni, Y.; Sültmann, H.; Mutz, P.; Hiet, M.S.; Vondran, F.W.R.; Bartenschlager, R.; Urban, S. Hepatitis D Virus Replication is Sensed by MDA5 and Induces IFN-β/λ Responses in Hepatocytes. J. Hepatol. 2018, 69, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hantz, O.; Parent, R.; Durantel, D.; Gripon, P.; Guguen-Guillouzo, C.; Zoulim, F. Persistence of the Hepatitis B Virus Covalently Closed Circular DNA in HepaRG Human Hepatocyte-like Cells. J. Gen. Virol. 2009, 90, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Claro Da Silva, T.; Polli, J.E.; Swaan, P.W. The Solute Carrier Family 10 (SLC10): Beyond Bile Acid Transport. Mol. Asp. Med. 2013, 34, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.U.T.; Seeger, C.; King, R.W. Inducible Expression of Human Hepatitis B Virus (HBV) in Stably Transfected Hepatoblastoma Cells: A Novel System for Screening Potential Inhibitors of HBV Replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Guan, W.; Sun, S.; Li, B.; Li, H.; Kang, F.; Kang, J.; Yang, D.; Nassal, M.; Sun, D. Stable Human Hepatoma Cell Lines for Efficient Regulated Expression of Nucleoside/Nucleotide Analog Resistant and Vaccine Escape Hepatitis B Virus Variants and Woolly Monkey Hepatitis B Virus. PLoS ONE 2015, 10, e0145746. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Yang, J.; Jo, E.; Park, K.H.P.; Kim, H.; Than, T.T.; Song, X.; Qi, X.; Dai, X.; Park, S.; et al. Efficient Long-Term Amplification of Hepatitis B Virus Isolates After Infection of Slow Proliferating HepG2-NTCP Cells. J. Hepatol. 2019, 71, 289–300. [Google Scholar] [CrossRef]
- Guo, J.-T.; Xu, X.; Block, T.M. Sulfamoylbenzamide Derivatives as Antiviral Agents Against HBV Infection. Patent WO 2013006394A1, 10 January 2013. [Google Scholar]
- George, D.H.; Osvaldo, A.F. Hepatitis B Antiviral Agents. Patent WO 2013096744A1, 27 June 2013. [Google Scholar]
- Last, S.J.; Raboisson, P.J.-M.B.; Rombouts, G.; Vandyck, K.; Verschueren, W.G. Sulfamoyl-Arylamides and the Use Thereof as Medicaments for the Treatment of Hepatitis B. Patent WO 2014033170A1, 6 March 2014. [Google Scholar]
- Vandyck, K.; Rombouts, G.; Stoops, B.; Tahri, A.; Vos, A.; Verschueren, W.; Wu, Y.; Yang, J.; Hou, F.; Huang, B.; et al. Synthesis and Evaluation of N-Phenyl-3-Sulfamoyl-Benzamide Derivatives as Capsid Assembly Modulators Inhibiting Hepatitis B Virus (HBV). J. Med. Chem. 2018, 61, 6247–6260. [Google Scholar] [CrossRef]
- Baugh, S.D.P.; Ye, H.; Xu, X.; Guo, J.-T. Novel Antiviral Agents against HBV Infection. Patent WO 2014106019A2, 3 July 2014. [Google Scholar]
- George, D.H.; Scott, K. Azepane Derivatives and Methods of Treating Hepatitis B Infections. Patent WO 2015109130A1, 23 July 2015. [Google Scholar]
- Kuduk, S.D.; Lam, A.M.; Espiritu, C.; Vogel, R.; Lau, V.; Klumpp, K.; Flores, O.A.; Hartman, G.D. SAR Studies in the Sulfonyl Carboxamide Class of HBV Capsid Assembly Modulators. Bioorg. Med. Chem. Lett. 2019, 29, 2405–2409. [Google Scholar] [CrossRef] [PubMed]
- Sari, O.; Boucle, S.; Cox, B.D.; Ozturk, T.; Russell, O.O.; Bassit, L.; Amblard, F.; Schinazi, R.F. Synthesis of Sulfamoylbenzamide Derivatives as HBV Capsid Assembly Effector. Eur. J. Med. Chem. 2017, 138, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Liu, L.; Li, Y.; Luo, H. Sulfamide Aryl Formamide Derivative and Preparation Method Therefore and Uses Thereof. Patent WO 2019206072A1, 31 October 2019. [Google Scholar]
- Ivachtchenko, A.V.; Ivachtchenko, A.A.; Savchuk, N.F. Hepatitis B Virus (HBV) Inhibitor. Patent WO 2019/017814 A1, 24 January 2019. [Google Scholar]
- Raffaele, D.F.; Adolfo, P.; Pietro, R. Inhibitors of Hepatitis B Virus. Patent WO 2020016427A1, 23 January 2020. [Google Scholar]
- Lv, K.; Wu, S.; Li, W.; Geng, Y.; Wu, M.; Zhou, J.; Li, Y.; Gao, Q.; Liu, M. Design, Synthesis and Anti-HBV Activity of NVR3-778 Derivatives. Bioorg. Chem. 2020, 94, 103363. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; Li, W.; Wu, S.; Geng, Y.; Wang, A.; Yang, L.; Huang, M.; Chowdhury, K.; Li, Y.; Liu, M. Amino Acid Prodrugs of NVR 3-778: Design, Synthesis and Anti-HBV Activity. Bioorg. Med. Chem. Lett. 2020, 30, 127103. [Google Scholar] [CrossRef] [PubMed]
- Na, H.G.; Imran, A.; Kim, K.; Han, H.S.; Lee, Y.J.; Kim, M.-J.; Yun, C.-S.; Jung, Y.-S.; Lee, J.-Y.; Han, S.B. Discovery of a New Sulfonamide Hepatitis B Capsid Assembly Modulator. ACS Med. Chem. Lett. 2020, 11, 166–171. [Google Scholar] [CrossRef]
- Lee, Y.H.; Cha, H.M.; Hwang, J.Y.; Park, S.Y.; Vishakantegowda, A.G.; Imran, A.; Lee, J.Y.; Yi, Y.S.; Jun, S.; Kim, G.H.; et al. Sulfamoylbenzamide-Based Capsid Assembly Modulators for Selective Inhibition of Hepatitis B Viral Replication. ACS Med. Chem. Lett. 2021, 12, 242–248. [Google Scholar] [CrossRef]
- Wang, S.; Ren, Y.; Li, Q.; Wang, Y.; Jiang, X.; Xu, S.; Zhang, X.; Zhao, S.; Bradley, D.P.; Woodson, M.E.; et al. Design, Synthesis, and Biological Evaluation of Novel Sulfamoylbenzamide Derivatives as HBV Capsid Assembly Modulators. Bioorg. Chem. 2022, 129, 106192. [Google Scholar] [CrossRef] [PubMed]
- Xiangkai, J.; Xiangyi, J.; Jiang, C.K.; Yujie, R.; Lide, H.; Zhen, G.; Dongwei, K.R.J.; Zhang, X.S.Z.; Zhao, K.W.; Xinyong, L.P.Z. Design, Synthesis, and Evaluation of a Set of Carboxylic Acid and Phosphate Prodrugs Derived from HBV Capsid Protein Allosteric Modulator NVR 3-778. Molecules 2022, 27, 5987. [Google Scholar] [CrossRef]
- Liu, H.; Okazaki, S.; Shinoda, W. Heteroaryldihydropyrimidines Alter Capsid Assembly by Adjusting the Binding Affinity and Pattern of the Hepatitis B Virus Core Protein. J. Chem. Inf. Model. 2019, 59, 5104–5110. [Google Scholar] [CrossRef]
- Vandyck, K.; Hache, G.Y.P.; Last, S.J.; Raboisson, P.J.M.B. N-Phenyl-Carboxamide Derivatives and the Use Thereof as Medicaments for the Treatment of Hepatitis B. Patent WO 2014161888A1, 9 October 2014. [Google Scholar]
- Hartman, G.D. Hepatitis B Antiviral Agents. U.S. Patent US20140275167A1, 18 September 2014. [Google Scholar]
- Wang, Z.; Fan, G.; Lu, C.; Yang, S.; Wang, X. Substituted Pyrrolizines for the Treatment of Hepatitis B. U.S. Patent 11591334B2, 28 February 2023. [Google Scholar]
- Wang, Z.; Fan, G.; Lu, C.; Yang, S.; Wang, X. Bicyclic Nucleocapsid Inhibitor and Use of Same as Drug in Treatment of Hepatitis B. World Organization Patent WO2018202155A1, 8 November 2018. [Google Scholar]
- Wang, Z.; Zeng, Z. Bi-heterocyclic Nucleocapsid Inhibitor and Pharmaceutical Use Thereof. Patent WO 2019210879A1, 7 November 2019. [Google Scholar]
- Liu, L.; Wang, M.; Li, C.; Han, X.; Xie, Y.; Feng, K.; Zhang, L.; Chen, Y.; Jia, H. Design, Synthesis and Biological Evaluation of Novel Dihydrobenzodioxine Derivatives as HBV Capsid Protein Inhibitors. Bioorg. Chem. 2022, 128, 106052. [Google Scholar] [CrossRef]
- Yin, J.; Feng, Z.; Li, Z.; Hu, J.; Hu, Y.; Cai, X.; Zhou, H.; Wang, K.; Tang, N.; Huang, A.; et al. Synthesis and Evaluation of N-Sulfonylpiperidine-3-Carboxamide Derivatives as Capsid Assembly Modulators Inhibiting HBV in Vitro and in HBV-Transgenic Mice. Eur. J. Med. Chem. 2023, 249, 115141. [Google Scholar] [CrossRef] [PubMed]
- Vandyck, K.; Hache, G.Y.P.; Last, S.J.; McGowan, D.C. Sulphamoylpyrrolamide Derivatives and the Use Thereof as Medicaments for the Treatment of Hepatitis B. Patent WO 2014184350A1, 20 November 2014. [Google Scholar]
- Chen, A.; Bravo, Y.; Stock, N.; Pedram, B. Sulfide Alkyl and Pyridyl Reverse Sulfonamide Compounds for HBV Treatment. Patent WO 2016089990A1, 9 June 2016. [Google Scholar]
- Qiu, Y.L.; Li, W.; Cao, H.; Jin, M. Hepatitis B Antiviral Agents. Patent WO 2016183266A1, 17 November 2016. [Google Scholar]
- Yinsheng, Z.; Wangwei, A.; Hangzhou, S.; Yuan, L. Capsid Protein Assembly Inhibitor, and Pharmaceutical Composition and Use Thereof. Patent WO 2019154343A1, 15 August 2019. [Google Scholar]
- Vendeville, S.M.R.; Last, S.J.; Demin, S.D.; Grosse, S.C. Cyclized Sulfamoylarylamide Derivatives and the Use Thereof as Medicaments for the Treatment of Hepatitis B. Patent WO 2017001655A1, 5 July 2017. [Google Scholar]
- Cole, A.G.; Dorsay, B.D.; Kakarla, R.; Kultgen, S.; Quintero, J. Substituted Dihydroindene-4-Carboxamides and Analogs Thereof, and Methods Using Same. Patent WO 2018172852A1, 27 September 2018. [Google Scholar]
- Wang, Z.; Wang, X.; Fan, G.; Lu, C. Cyclothiourea Compound and Use Thereof. Patent WO 2018133846A1, 26 July 2018. [Google Scholar]
- Wang, Z.; Wang, X.; Yang, S.; Zeng, Z. Endocyclic Thiamidinoamide-Arylamide Compound and Use Thereof for Treating Hepatitis B. U.S. Patent 11168055B2, 9 November 2021. [Google Scholar]
- Walker, M.; Li, L.; Simon, N. Cyclic Sulfamide Compounds for Treatment of HBV. Patent WO 2020051319A1, 12 March 2020. [Google Scholar]
- Walker, M.; Li, L.; Simon, N. Cyclic Sulfamide Compounds for Treatment of HBV. Patent WO 2020051320A1, 12 March 2020. [Google Scholar]
- Beigelman, L.; Smith, D.S. Bicyclic Sulfonamides. Patent WO 2020167984A1, 20 August 2020. [Google Scholar]
- Raffaele, D.F.; Lorena, D.; Luca, G.; Matteo, I. Cyclic Inhibitors of Hepatitis B Virus. Patent WO 2020016434, 23 January 2020. [Google Scholar]
- Raffaele, D.F.; Lorena, D.; Luca, G.; Matteo, I. Tricyclic Inhibitors of Hepatitis B Virus. Patent WO 2020030781A1, 13 February 2020. [Google Scholar]
- Raffaele, D.F.; Lorena, D.; Luca, G.; Matteo, I. Oxalamido-Substituted Tricyclic Inhibitors of Hepatitis B Virus. Patent WO 2020234483A1, 26 November 2020. [Google Scholar]
- Wang, C.; Pei, Y.; Pei, Y.; Wang, L.; Li, S.; Jiang, C.; Tan, X.; Dong, Y.; Xiang, Y.; Ma, Y.; et al. Discovery of (1 H-Pyrazolo[3,4-c]Pyridin-5-Yl)Sulfonamide Analogues as Hepatitis B Virus Capsid Assembly Modulators by Conformation Constraint. J. Med. Chem. 2020, 63, 6066–6089. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrails.gov. Available online: https://clinicaltrials.gov (accessed on 21 September 2023).
- Hepatitis B Foundation. Available online: http://www.hepb.org/treatment-and-management/drug-watch/ (accessed on 21 September 2023).
- Pan, Y.; Xia, H.; He, Y.; Zeng, S.; Shen, Z.; Huang, W. The Progress of Molecules and Strategies for the Treatment of HBV Infection. Front. Cell. Infect. Microbiol. 2023, 13, 1128807. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ko, C.; Lee, J.Y.; Kim, M. Current Progress in the Development of Hepatitis b Virus Capsid Assembly Modulators: Chemical Structure, Mode-of-Action and Efficacy. Molecules 2021, 26, 7420. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Gao, L.; Han, X.; Hu, T.; Hu, Y.; Liu, H.; Thomas, A.W.; Yan, Z.; Yang, S.; Young, J.A.T.; et al. Discovery of Small Molecule Therapeutics for Treatment of Chronic HBV Infection. ACS Infect. Dis. 2018, 4, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Nijampatnam, B.; Liotta, D.C. Recent Advances in the Development of HBV Capsid Assembly Modulators. Curr. Opin. Chem. Biol. 2019, 50, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, F.; Tong, X.; Hoffmann, D.; Zuo, J.; Lu, M. Treatment of Chronic Hepatitis B Virus Infection Using Small Molecule Modulators of Nucleocapsid Assembly: Recent Advances and Perspectives. ACS Infect. Dis. 2019, 5, 713–724. [Google Scholar] [CrossRef]
- Viswanathan, U.; Mani, N.; Hu, Z.; Ban, H.; Du, Y.; Hu, J.; Chang, J.; Guo, J.T. Targeting the Multifunctional HBV Core Protein as a Potential Cure for Chronic Hepatitis B. Antiviral Res. 2020, 182, 104917. [Google Scholar] [CrossRef]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Yuen Chan, H.L.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7. [Google Scholar] [CrossRef]
- Tavis, J.E.; Lomonosova, E. NVR 3-778 Plus Pegylated Interferon-α Treatment for Chronic Hepatitis B Viral Infections: Could 1 + 1 = 3? Gastroenterology 2018, 154, 481–482. [Google Scholar] [CrossRef]
- Lenz, O.; Verbinnen, T.; Hodari, M.; Talloen, W.; Vandenbossche, J.; Shukla, U.; Berby, F.; Scholtes, C.; Blue, D.; Yogaratnam, J.; et al. HBcrAg Decline in JNJ-56136379-Treated Chronic Hepatitis B Patients: Results of a Phase 1 Study. J. Hepatol. 2019, 70, e473–e474. [Google Scholar] [CrossRef]
- Zoulim, F.; Lenz, O.; Vandenbossche, J.J.; Talloen, W.; Verbinnen, T.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; Crespo, J.; et al. JNJ-56136379, an HBV Capsid Assembly Modulator, is Well-Tolerated and has Antiviral Activity in a Phase 1 Study of Patients with Chronic Infection. Gastroenterology 2020, 159, 521–533.e9. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.; Hou, J.; Asselah, T.; Chan, H.; Zoulim, F.; Tanaka, Y.; Janczewska, E.; Nahass, R.; Bourgeois, S.; Buti, M.; et al. Efficacy and Safety Results of the Phase 2 JNJ-56136379 JADE Study in Patients with Chronic Hepatitis B: Interim Week 24 Data. J. Hepatol. 2020, 73, S129–S130. [Google Scholar] [CrossRef]
- Gane, E.; Yuen, M.F.; Kakuda, T.N.; Ogawa, T.; Takahashi, Y.; Goeyvaerts, N.; Lonjon-Domanec, I.; Vaughan, T.; Schluep, T.; Hamilton, J.; et al. JNJ-73763989 Pharmacokinetics and Safety: Liver-Targeted SiRNAs against Hepatitis B Virus, in Japanese and Non-Japanese Healthy Adults, and Combined with JNJ-56136379 and a Nucleos(t)ide Analogue in Patients with Chronic Hepatitis B. Antivir. Ther. 2022, 27, 13596535221093856. [Google Scholar] [CrossRef]
- Qi, H.; Dawei, C.; Ran, Y.; Lichun, L.; Yahua, Z.; Lida, G.; Alexebdre, M.; Yi, Z.; Ariel, T.; Kirk, H.; et al. Preclinical Profile and Characterization of the Hepatitis B Virus Core Protein Inhibitor ABI-H0731. Antimicrob. Agents Chemother. 2020, 64, e01463-20. [Google Scholar] [CrossRef]

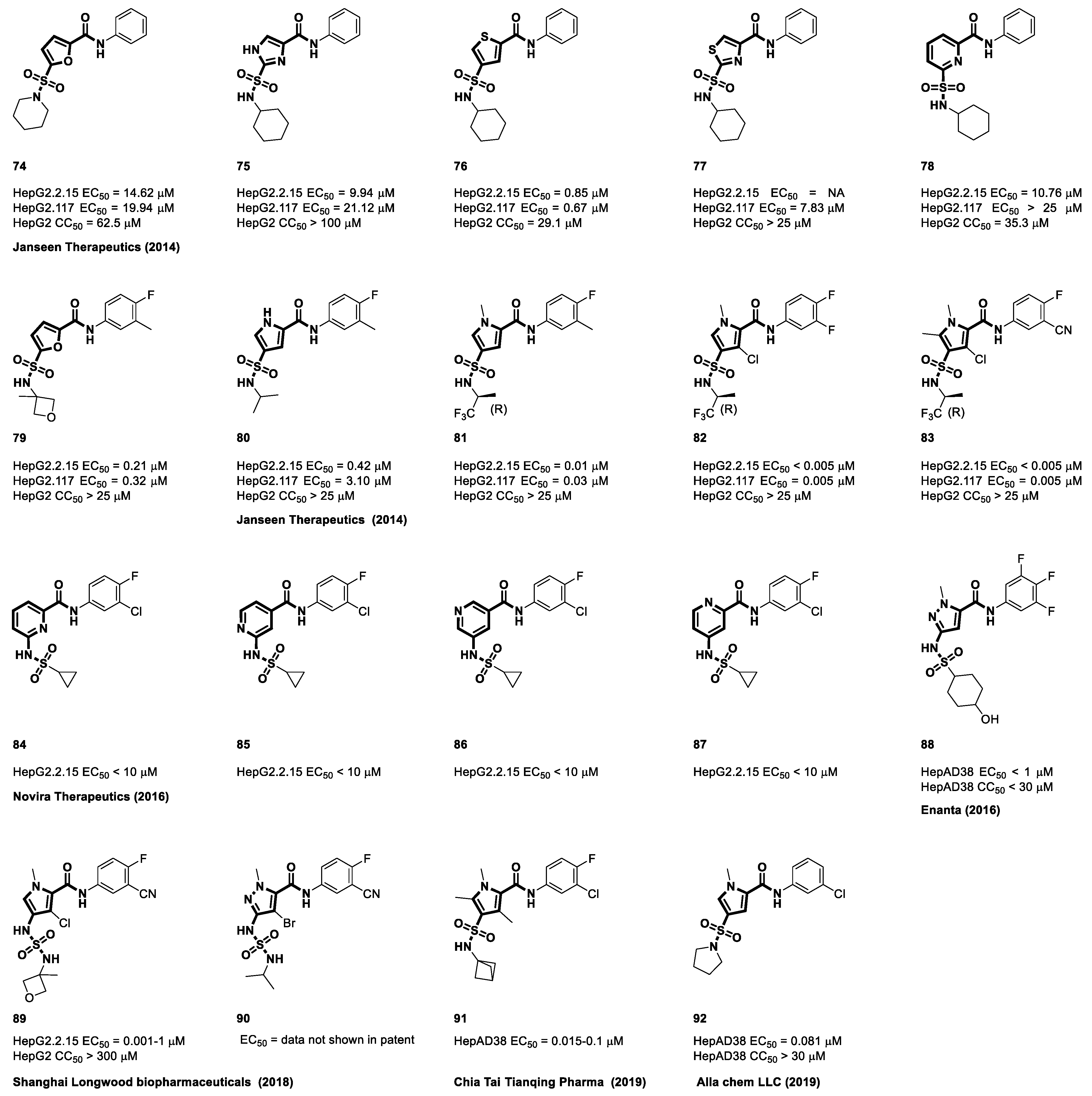
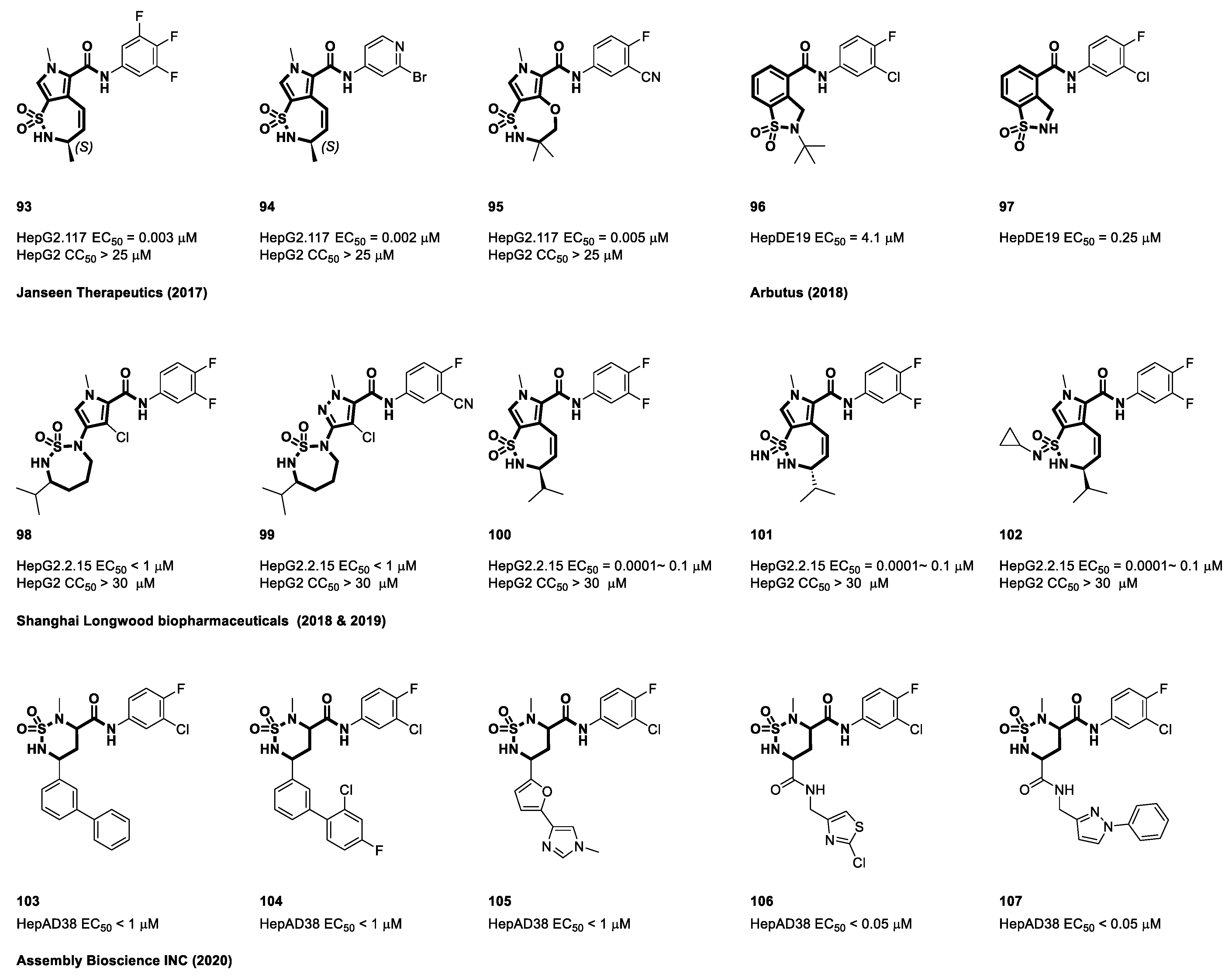

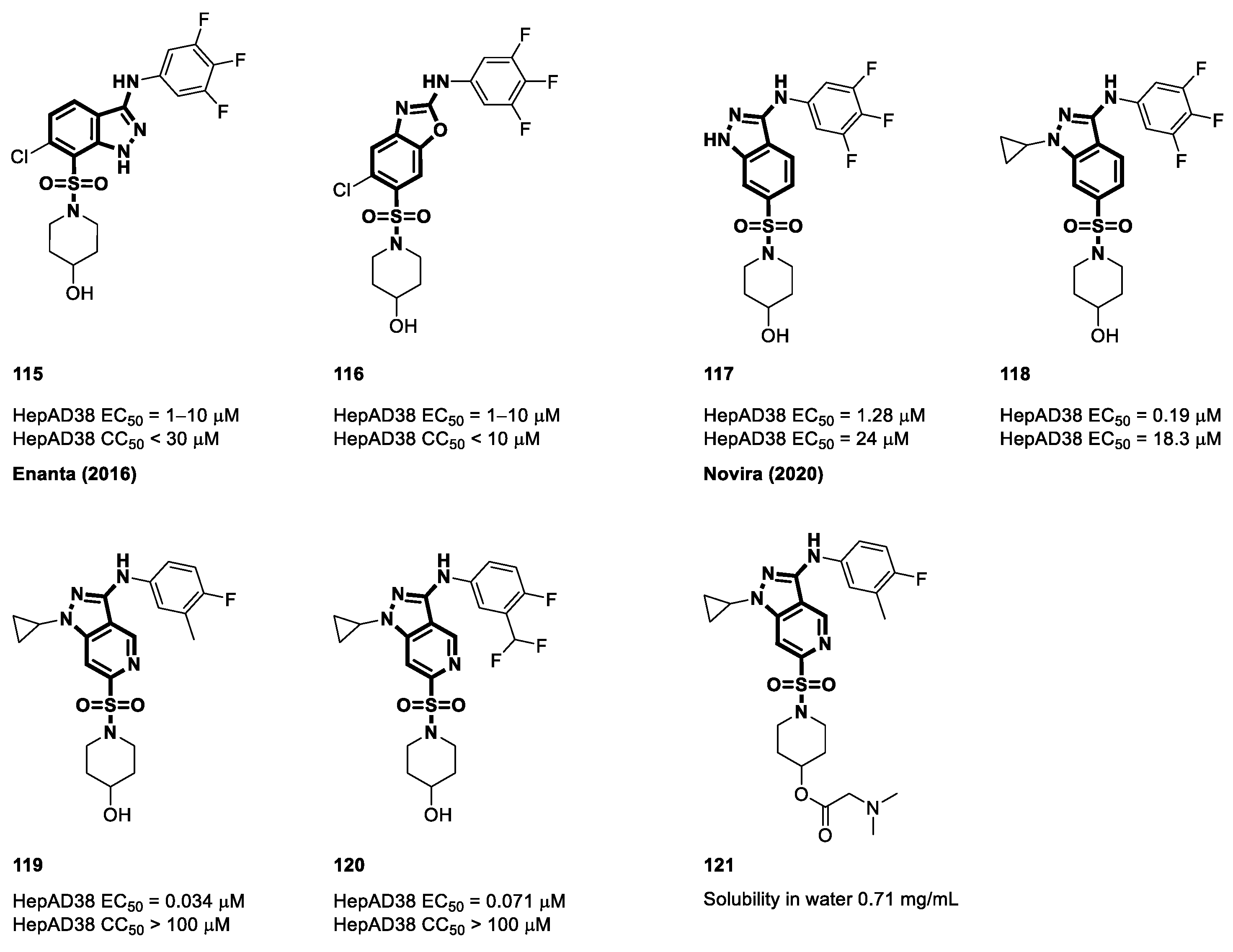


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Culture Models | Pros and Cons of Cell Culture Models | |||
|---|---|---|---|---|
| Entry | Replication | Secretion | De Novo Spread | |
| PHHs | high | high | low | low |
| HepaRG | low | high | low | no |
| HuH7 | no | moderate | moderate | no |
| HuH7-NTCP | high | moderate | low | no |
| HepAD38/HepG2.2.15/HepG2.117 | no | high | high | no |
| HepG2-NTCP | high | high | low | no |
| HepG2-NTCP sec+ | high | high | moderate | moderate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayak, S.; Gowda, J.; Abbas, S.A.; Kim, H.; Han, S.B. Recent Advances in the Development of Sulfamoyl-Based Hepatitis B Virus Nucleocapsid Assembly Modulators. Viruses 2023, 15, 2367. https://doi.org/10.3390/v15122367
Nayak S, Gowda J, Abbas SA, Kim H, Han SB. Recent Advances in the Development of Sulfamoyl-Based Hepatitis B Virus Nucleocapsid Assembly Modulators. Viruses. 2023; 15(12):2367. https://doi.org/10.3390/v15122367
Chicago/Turabian StyleNayak, Sandesha, Jayaraj Gowda, Syed Azeem Abbas, Hyejin Kim, and Soo Bong Han. 2023. "Recent Advances in the Development of Sulfamoyl-Based Hepatitis B Virus Nucleocapsid Assembly Modulators" Viruses 15, no. 12: 2367. https://doi.org/10.3390/v15122367
APA StyleNayak, S., Gowda, J., Abbas, S. A., Kim, H., & Han, S. B. (2023). Recent Advances in the Development of Sulfamoyl-Based Hepatitis B Virus Nucleocapsid Assembly Modulators. Viruses, 15(12), 2367. https://doi.org/10.3390/v15122367