
Dramatic Differences between the Structural Susceptibility of the S1 Pre- and S2 Postfusion States of the SARS-CoV-2 Spike Protein to External Electric Fields Revealed by Molecular Dynamics Simulations

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Simulation Setup
2.2. Principal-Component-Analysis
2.3. Free-Energy Landscape Estimation
2.4. MD Analysis
2.5. Electrostatic Potential Surface Calculations
3. Results
3.1. Global Structural Changes in the S1 and S2 Subunits of the Spike Protein under Electric Fields

3.2. Effects of Moderate Electric Fields on the Secondary Structure of the Receptor Binding Domain

3.3. Stability of the Field-Induced Final Conformational States in the RDB Spike Protein
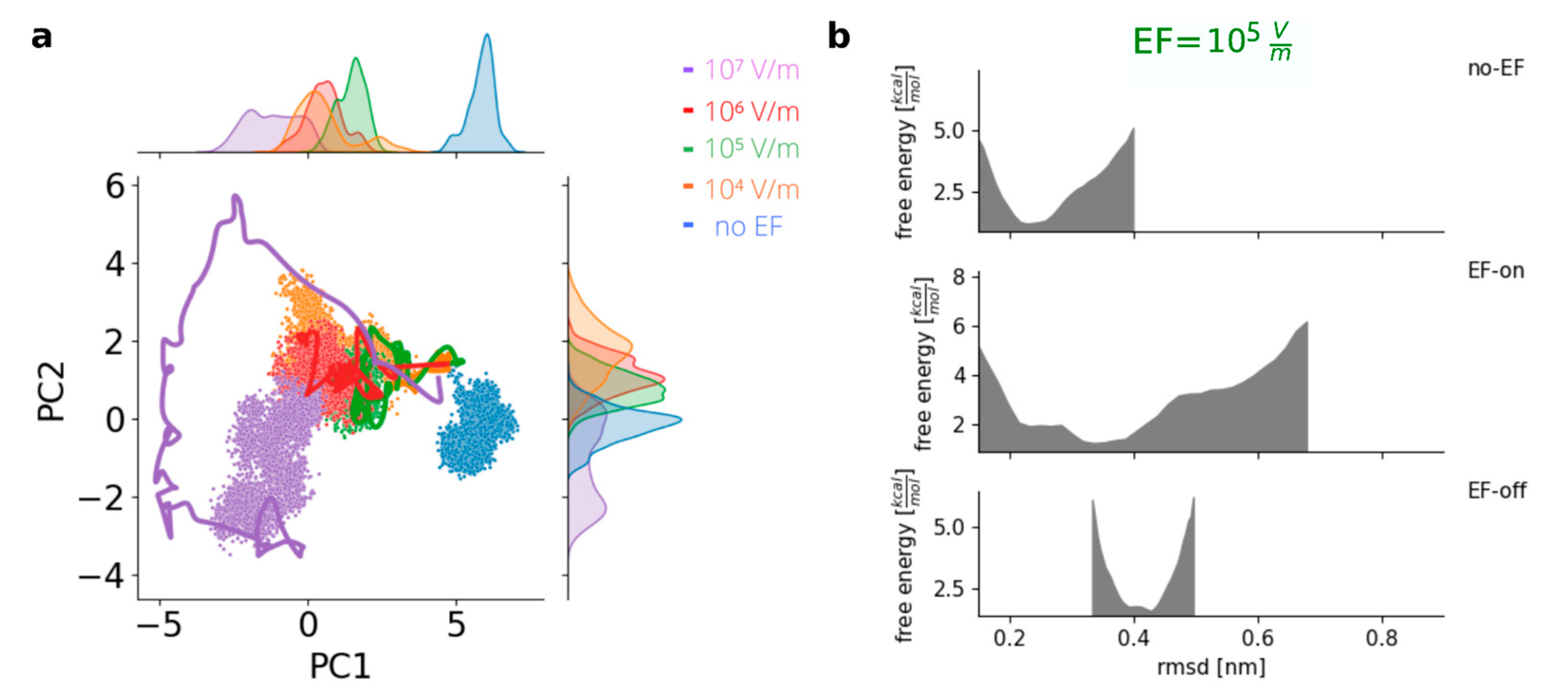
3.4. Disruption of the Charge Complementarity between RBD and ACE2 upon the Application of an Electric Field

3.5. Influence of Electric Fields on the Fusion Core Region of the S2 Subunit of the Spike Protein
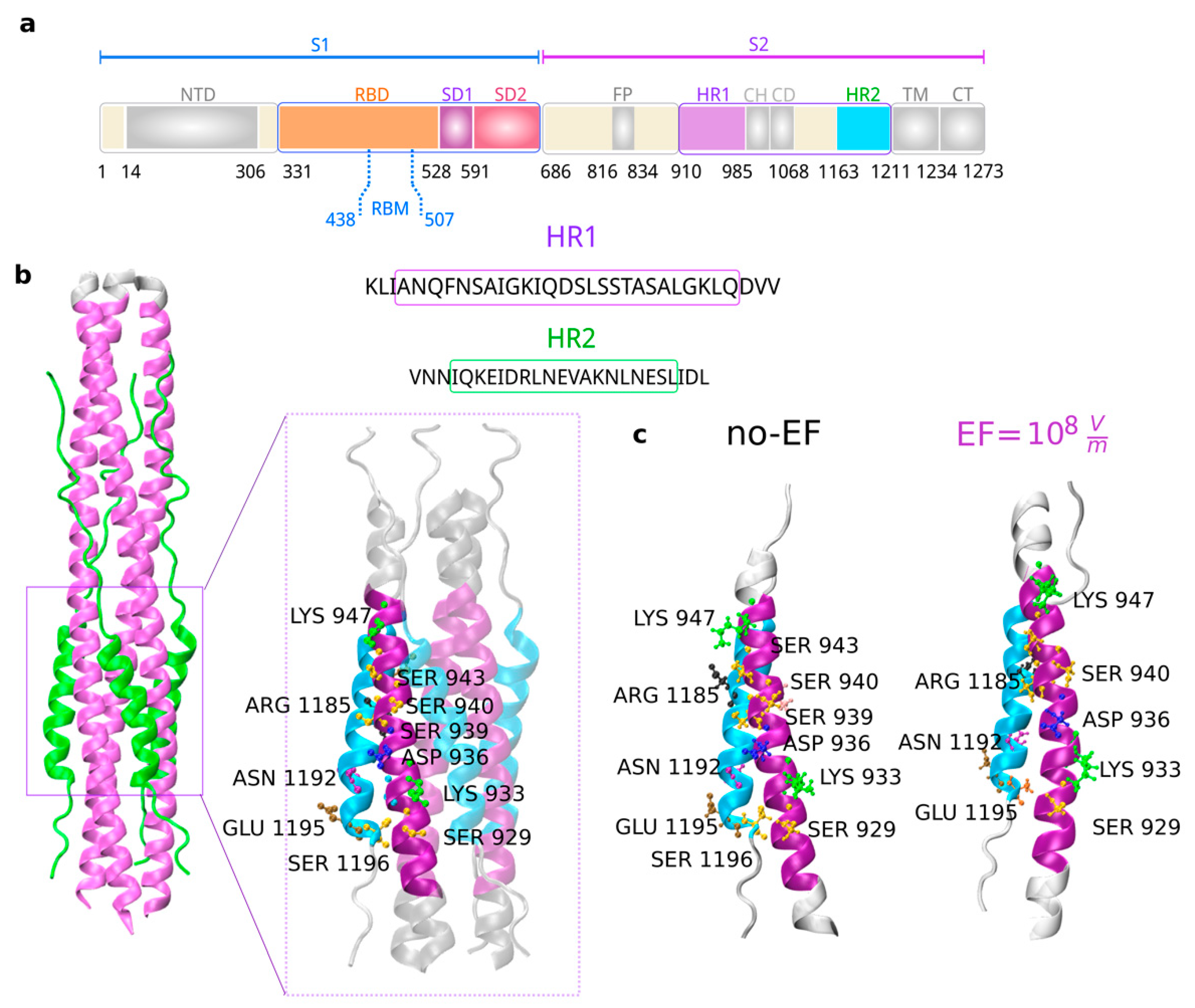
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- About COVID-19|Variants of the Virus—Center for Disease Control and Prevention. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/ (accessed on 30 September 2023).
- Tulimilli, S.V.; Dallavalasa, S.; Basavaraju, C.G.; Kumar Rao, V.; Chikkahonnaiah, P.; Madhunapantula, S.V.; Veeranna, R.P. Variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and vaccine effectiveness. Vaccines 2022, 10, 1751. [Google Scholar] [CrossRef] [PubMed]
- Pipitò, L.; Reynolds, C.A.; Mobarec, J.C.; Vickery, O.; Deganutti, G. A Pathway Model to Understand the Evolution of Spike Protein Binding to ACE2 in SARS-CoV-2 Variants. Biomolecules 2022, 12, 1607. [Google Scholar] [CrossRef]
- Colman, P.M.; Lawrence, M.C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003, 4, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Benhaim, M.A.; Lee, K.K. New biophysical approaches reveal the dynamics and mechanics of type I viral fusion machinery and their interplay with membranes. Viruses 2020, 12, 413. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Ng, K.T.; Mohd-Ismail, N.K.; Tan, Y.J. Spike S2 subunit: The dark horse in the race for prophylactic and therapeutic interventions against SARS-CoV-2. Vaccines 2021, 9, 178. [Google Scholar] [CrossRef]
- Xia, X. Domains and functions of spike protein in Sars-Cov-2 in the context of vaccine design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479, 498–507. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Ranjan, A. The metastable states of proteins. Protein Sci. 2020, 29, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Pre-fusion structure of a human coronavirus spike protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, S.H.; Lee, M.Y.; Yu, M.H. Regulation of protein function by native metastability. Proc. Natl. Acad. Sci. USA 2000, 97, 7727–7731. [Google Scholar] [CrossRef]
- Baker, D.; Agard, D.A. Kinetics versus thermodynamics in protein folding. Biochemistry 1994, 33, 7505–7509. [Google Scholar] [CrossRef]
- Peng, R.; Wu, L.A.; Wang, Q.; Qi, J.; Gao, G.F. Cell entry by SARS-CoV-2. Trends Biochem. Sci. 2021, 46, 848–860. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Hekstra, D.R.; White, K.I.; Socolich, M.A.; Henning, R.W.; Šrajer, V.; Ranganathan, R. Electric-field-stimulated protein mechanics. Nature 2016, 540, 400–405. [Google Scholar]
- Ojeda-May, P.; Garcia, M.E. Electric field-driven disruption of a native β-sheet protein conformation and generation of a helix-structure. Biophys. J. 2010, 99, 595–599. [Google Scholar] [CrossRef]
- Zhang, Q.; Shao, D.; Xu, P.; Jiang, Z. Effects of an electric field on the conformational transition of the protein: Pulsed and oscillating electric fields with different frequencies. Polymers 2021, 14, 123. [Google Scholar] [CrossRef]
- Urabe, G.; Katagiri, T.; Katsuki, S. Intense pulsed electric fields denature urease protein. Bioelectricity 2020, 2, 33–39. [Google Scholar] [CrossRef]
- Baumketner, A. Electric field as a disaggregating agent for amyloid fibrils. J. Phys. Chem. B 2014, 118, 14578–14589. [Google Scholar] [CrossRef] [PubMed]
- De Biase, P.M.; Paggi, D.A.; Doctorovich, F.; Hildebrandt, P.; Estrin, D.A.; Murgida, D.H.; Marti, M.A. Molecular basis for the electric field modulation of cytochrome c structure and function. J. Am. Chem. Soc. 2009, 131, 16248–16256. [Google Scholar] [CrossRef]
- Arbeitman, C.R.; Rojas, P.; Ojeda-May, P.; Garcia, M.E. The SARS-CoV-2 spike protein is vulnerable to moderate electric fields. Nat. Commun. 2021, 12, 5407. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. Available online: https://www.rcsb.org/ (accessed on 15 July 2023). [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Nguyen, P.H.; Stock, G. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins Struct. Funct. Bioinform. 2005, 58, 45–52. [Google Scholar] [CrossRef]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef]
- Fabian, P. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825. [Google Scholar]
- Calvo, F. Sampling along reaction coordinates with the Wang-Landau method. Mol. Phys. 2002, 100, 3421–3427. [Google Scholar] [CrossRef]
- Adjanor, G.; Athenes, M.; Calvo, F. Free energy landscape from path-sampling: Application to the structural transition in LJ 38. Eur. Phys. J. B Condens. Matter Complex Syst. 2006, 53, 47–60. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: Cambridge, MA, USA, 2002; ISBN 0-12-267351-4. [Google Scholar]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32 (Suppl. S2), W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Spiga, O.; Bernini, A.; Ciutti, A.; Chiellini, S.; Menciassi, N.; Finetti, F.; Causarono, V.; Anselmi, F.; Prischi, F.; Niccolai, N. Molecular modelling of S1 and S2 subunits of SARS coronavirus spike glycoprotein. Biochem. Biophys. Res. Commun. 2003, 310, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H. Crystallographic and biophysical analysis of the fusion core from SARS-CoV-2 spike protein. J. Chin. Chem. Soc. 2023, 70, 1208–1218. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Spinello, A.; Saltalamacchia, A.; Magistrato, A. Is the rigidity of SARS-CoV-2 spike receptor-binding motif the hallmark for its enhanced infectivity? Insights from all-atom simulations. J. Phys. Chem. Lett. 2020, 11, 4785–4790. [Google Scholar] [CrossRef]
- Baral, P.; Bhattarai, N.; Hossen, M.L.; Stebliankin, V.; Gerstman, B.S.; Narasimhan, G.; Chapagain, P.P. Mutation-induced changes in the receptor-binding interface of the SARS-CoV-2 Delta variant B. 1.617. 2 and implications for immune evasion. Biochem. Biophys. Res. Commun. 2021, 574, 14–19. [Google Scholar] [CrossRef]
- Pitsillou, E.; Liang, J.J.; Beh, R.C.; Hung, A.; Karagiannis, T. Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain. Comput. Biol. Med. 2022, 149, 106035. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Gilson, M.K. Theory of electrostatic interactions in macromolecules. Curr. Opin. Struct. Biol. 1995, 5, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Socher, E.; Heger, L.; Paulsen, F.; Zunke, F.; Arnold, P. Molecular dynamics simulations of the delta and omicron SARS-CoV-2 spike–ACE2 complexes reveal distinct changes between both variants. Comput. Struct. Biotechnol. J. 2022, 20, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Sang, P.; Chen, Y.Q.; Liu, M.T.; Wang, Y.T.; Yue, T.; Li, Y.; Yin, Y.R.; Yang, L.Q. Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants. Int. J. Mol. Sci. 2022, 23, 14796. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 3618. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K.; Yang, M.; Li, Y.; Zhu, L.; Wang, N.; Lv, Z.; et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 2020, 369, 77–81. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef]
- Xiong, D.; Zhao, X.; Luo, S.; Duan, L. Insights from computational analysis: How does the SARS-CoV-2 Delta (B. 1.617. 2) variant hijack ACE2 more effectively? Phys. Chem. Chem. Phys. 2022, 24, 8683–8694. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The global epidemic of the SARS-CoV-2 delta variant, key spike mutations and immune escape. Front. Immunol. 2021, 12, 751778. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipskij, A.; Arbeitman, C.; Rojas, P.; Ojeda-May, P.; Garcia, M.E. Dramatic Differences between the Structural Susceptibility of the S1 Pre- and S2 Postfusion States of the SARS-CoV-2 Spike Protein to External Electric Fields Revealed by Molecular Dynamics Simulations. Viruses 2023, 15, 2405. https://doi.org/10.3390/v15122405
Lipskij A, Arbeitman C, Rojas P, Ojeda-May P, Garcia ME. Dramatic Differences between the Structural Susceptibility of the S1 Pre- and S2 Postfusion States of the SARS-CoV-2 Spike Protein to External Electric Fields Revealed by Molecular Dynamics Simulations. Viruses. 2023; 15(12):2405. https://doi.org/10.3390/v15122405
Chicago/Turabian StyleLipskij, Alexander, Claudia Arbeitman, Pablo Rojas, Pedro Ojeda-May, and Martin E. Garcia. 2023. "Dramatic Differences between the Structural Susceptibility of the S1 Pre- and S2 Postfusion States of the SARS-CoV-2 Spike Protein to External Electric Fields Revealed by Molecular Dynamics Simulations" Viruses 15, no. 12: 2405. https://doi.org/10.3390/v15122405
APA StyleLipskij, A., Arbeitman, C., Rojas, P., Ojeda-May, P., & Garcia, M. E. (2023). Dramatic Differences between the Structural Susceptibility of the S1 Pre- and S2 Postfusion States of the SARS-CoV-2 Spike Protein to External Electric Fields Revealed by Molecular Dynamics Simulations. Viruses, 15(12), 2405. https://doi.org/10.3390/v15122405