
The Metagenomic Analysis of Viral Diversity in Colorado Potato Beetle Public NGS Data
 , , , , and
, , , , and
Abstract
:1. Introduction
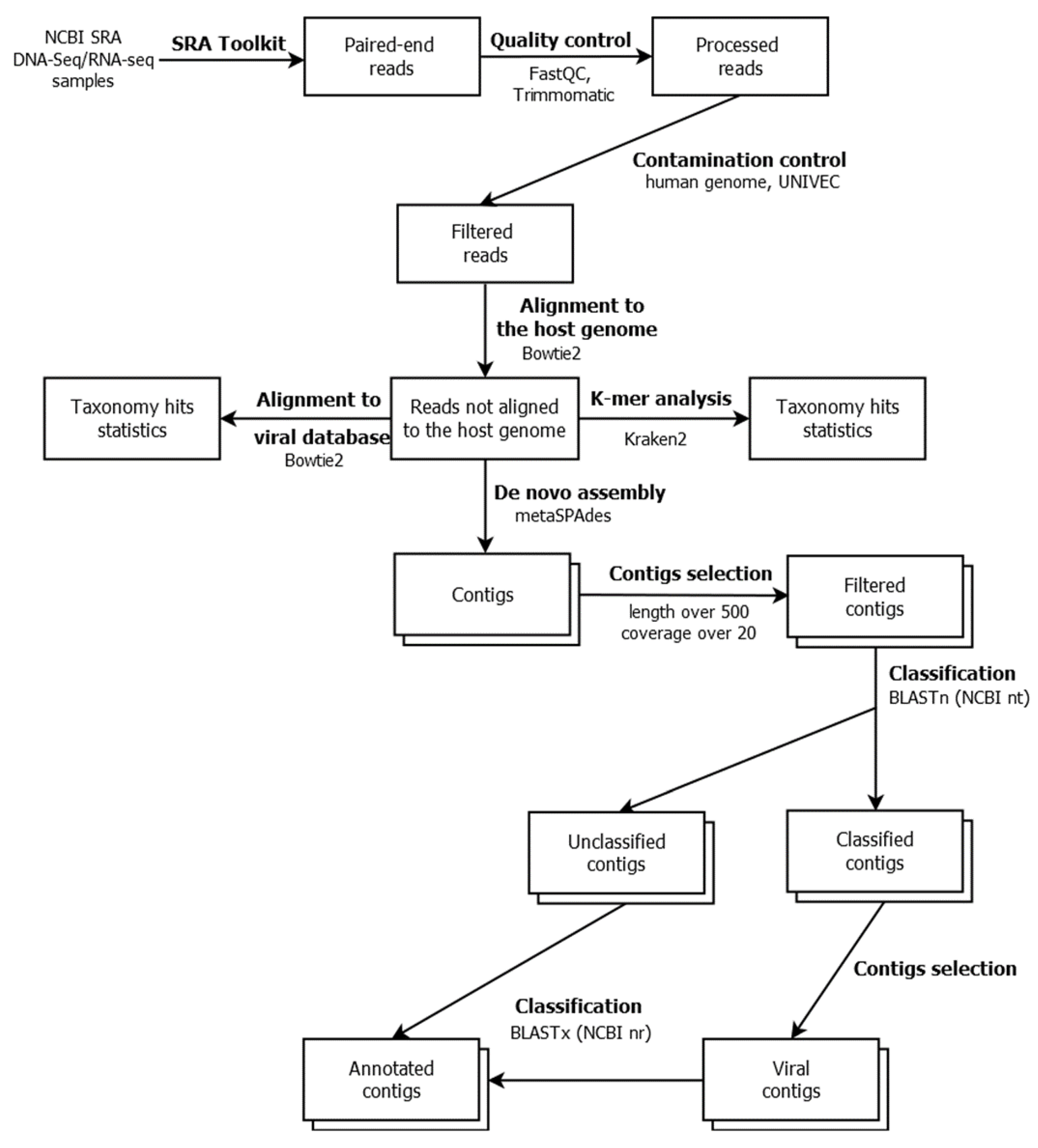
2. Materials and Methods
2.1. Genome and Transcriptome Data of Leptinotarsa Decemlineata Samples
2.2. Databases
- The NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra, accessed on June 2021).
- The BigViralDB—our custom database—combined nonredundant viral sequences assembled from four databases: NCBI RefSeq (11,566 viral sequences, June 2021) (https://www.ncbi.nlm.nih.gov/refseq/, accessed on June 2021), Virxicon (327,932 viral sequences, June 2021) [19], Virosaurus (823,421 viral sequences, April 2020) [20], and virus sequences from the NCBI Nucleotide database (1,367,485 viral sequences, June 2021). The database can be provided at request.
- The NCBI Genome Database (https://www.ncbi.nlm.nih.gov/genome/, accessed on July 2021).
- The NCBI nt database (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nt.gz, accessed on July 2021).
- The NCBI nr database (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz, accessed on July 2021).
- The NCBI UniVec database (https://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/, accessed on June 2021), a collection of unique vector sequences used in genetic engineering and biotechnology research (3137 sequences, June 2021).
2.3. Metagenome Assembly and Sequence Classification
2.4. Analysis of Reference Genomes of Selected Coleopterans
2.5. Biological Sample Collection and Preparation
2.6. Confirmation of the Presence of Bracovirus Genetic Fragments in the Genetic Material of L. decemlineata
3. Results
3.1. Analysis of the k-Mer Spectra
3.2. Contig Analysis
- Among the viral protein BLASTx hits for which the virus family was known, the best match was selected for each contig based on the e-value (minimum) and the bitscore (maximum)—2954 rows (Supplementary Table S4);
- Among the viral protein BLASTx hits for which the virus family was unknown, the best match was selected for each contig based on the e-value (minimum) and the bitscore (maximum)—1662 rows (Supplementary Table S5);
- Among the non-viral protein BLASTx hits, the best match was selected for each contig based on the e-value (minimum) and the bitscore (maximum)—13,252 rows (Supplementary Table S6).
3.3. Non-Viral Protein Hits Are Significantly Enriched with Unknown and Uncharacterized Proteins as Compared to Viral Protein Hits
3.4. Alignment Results
3.5. Analysis of Selected Coleopteran Species Reference Genomes to Detect Bracoviral Genetic Fragments
4. Discussion
4.1. Insect Viruses
4.2. Plant Viruses
4.3. Endogenous Virus Elements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bassi, C.; Guerriero, P.; Pierantoni, M.; Callegari, E.; Sabbioni, S. Novel Virus Identification through Metagenomics: A Systematic Review. Life 2022, 12, 2048. [Google Scholar] [CrossRef] [PubMed]
- Varghese, F.S.; van Rij, R.P. Insect Virus Discovery by Metagenomic and Cell Culture-Based Approaches. Methods Mol. Biol. 2018, 1746, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Breitbart, M. Exploring the Viral World through Metagenomics. Curr. Opin. Virol. 2011, 1, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Cingel, A.; Savić, J.; Lazarević, J.; Ćosić, T.; Raspor, M.; Smigocki, A.; Ninković, S. Extraordinary Adaptive Plasticity of Colorado Potato Beetle: “Ten-Striped Spearman” in the Era of Biotechnologicalwarfare. Int. J. Mol. Sci. 2016, 17, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sablon, L.; Dickens, J.C.; Haubruge, É.; Verheggen, F.J. Chemical Ecology of the Colorado Potato Beetle, Leptinotarsa decemlineata (Say) (Coleoptera: Chrysomelidae), and Potential for Alternative Control Methods. Insects 2012, 4, 31–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharijaya, A.; Vosman, B. Managing the Colorado Potato Beetle; the Need for Resistance Breeding. Euphytica 2015, 204, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Alyokhin, A.; Baker, M.; Mota-Sanchez, D.; Dively, G.; Grafius, E. Colorado Potato Beetle Resistance to Insecticides. Am. J. Potato Res. 2008, 85, 395–413. [Google Scholar] [CrossRef]
- Schoville, S.D.; Chen, Y.H.; Andersson, M.N.; Benoit, J.B.; Bhandari, A.; Bowsher, J.H.; Brevik, K.; Cappelle, K.; Chen, M.-J.M.; Childers, A.K.; et al. A Model Species for Agricultural Pest Genomics: The Genome of the Colorado Potato Beetle, Leptinotarsa decemlineata (Coleoptera: Chrysomelidae). Sci. Rep. 2018, 8, 1931. [Google Scholar] [CrossRef] [Green Version]
- Bale, J.; van Lenteren, J.; Bigler, F. Biological Control and Sustainable Food Production. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 761–776. [Google Scholar] [CrossRef] [Green Version]
- van Lenteren, J.C.; Bolckmans, K.; Köhl, J.; Ravensberg, W.J.; Urbaneja, A. Biological Control Using Invertebrates and Microorganisms: Plenty of New Opportunities. BioControl 2018, 63, 39–59. [Google Scholar] [CrossRef] [Green Version]
- Tanwar, R.S.; Dureja, P.; Rathore, H.S. Biopesticides; Elsevier Inc.: Amsterdam, The Netherlands, 2012; ISBN 9781439836255. [Google Scholar]
- Alyokhin, A.; Kryukov, V. Chapter 25—Ecology of a Potato Field. In Insect Pests of Potato, 2nd ed.; Alyokhin, A., Rondon, S.I., Gao, Y., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 451–462. ISBN 978-0-12-821237-0. [Google Scholar]
- Yu, Y.; Yuan, Y.; Gao, M. Construction of an Environmental Safe Bacillus thuringiensis Engineered Strain against Coleoptera. Appl. Microbiol. Biotechnol. 2016, 100, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
- Beas-Catena, A.; Sánchez-Mirón, A.; García-Camacho, F.; Contreras-Gómez, A.; Molina-Grima, E. Baculovirus Biopesticides: An Overview. J. Anim. Plant Sci. 2014, 24, 362–373. [Google Scholar]
- Ros, V.I.D.; Panziera, D.; Nalcacioglu, R.; Petersen, J.M.; Ryabov, E.; van Oers, M.M. Viral Diseases of Insects. In Invertebrate Pathology; Rowley, A.F., Coates, C.J., Whitten, M.W., Eds.; Oxford University Press: Oxford, UK, 2022; pp. 249–285. [Google Scholar] [CrossRef]
- Jones, R.A.C. Plant and Insect Viruses in Managed and Natural Environments: Novel and Neglected Transmission Pathways, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 101. [Google Scholar]
- Selman, B.J. Viruses and Chrysomelidae. In Biology of Chrysomelidae; Jolivet, P., Petitpierre, E., Hsiao, T.H., Eds.; Series Entomologica; Springer: Dordrecht, The Netherlands, 1988; Volume 42, pp. 379–387. [Google Scholar] [CrossRef]
- Kumar, A.; Congiu, L.; Lindström, L.; Piiroinen, S.; Vidotto, M.; Grapputo, A. Sequencing, de Novo Assembly and Annotation of the Colorado Potato Beetle, Leptinotarsa decemlineata, Transcriptome. PLoS ONE 2014, 9, e86012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudla, M.; Gutowska, K.; Synak, J.; Weber, M.; Bohnsack, K.S.; Lukasiak, P.; Villmann, T.; Blazewicz, J.; Szachniuk, M. Virxicon: A Lexicon of Viral Sequences. Bioinformatics 2020, 36, 5507–5513. [Google Scholar] [CrossRef] [PubMed]
- Gleizes, A.; Laubscher, F.; Guex, N.; Iseli, C.; Junier, T.; Cordey, S.; Fellay, J.; Xenarios, I.; Kaiser, L.; Le Mercier, P. Virosaurus a Reference to Explore and Capture Virus Genetic Diversity. Viruses 2020, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive Analysis of Metagenomics Data for Microbiome Studies and Pathogen Identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotskaya, U.N.; Kryukov, V.Y.; Kosman, E.; Tyurin, M.; Glupov, V.V. Identification of the Ricin-b-Lectin Ldrblk in the Colorado Potato Beetle and an Analysis of Its Expression in Response to Fungal Infections. J. Fungi 2021, 7, 364. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, V.Y.; Kabilov, M.R.; Smirnova, N.; Tomilova, O.G.; Tyurin, M.V.; Akhanaev, Y.B.; Polenogova, O.V.; Danilov, V.P.; Zhangissina, S.K.; Alikina, T.; et al. Bacterial Decomposition of Insects Post-Metarhizium Infection: Possible Influence on Plant Growth. Fungal Biol. 2019, 123, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Polenogova, O.V.; Noskov, Y.A.; Yaroslavtseva, O.N.; Kryukova, N.A.; Alikina, T.; Klementeva, T.N.; Andrejeva, J.; Khodyrev, V.P.; Kabilov, M.R.; Kryukov, V.Y.; et al. Influence of Bacillus thuringiensis and Avermectins on Gut Physiology and Microbiota in Colorado Potato Beetle: Impact of Enterobacteria on Susceptibility to Insecticides. PLoS ONE 2021, 16, e0248704. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Cholleti, H.; Hayer, J.; Fafetine, J.; Berg, M.; Blomström, A.L. Genetic Characterization of a Novel Picorna-like Virus in Culex spp. Mosquitoes from Mozambique. Virol. J. 2018, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hunnicutt, L.E.; Mozoruk, J.; Hunter, W.B.; Crosslin, J.M.; Cave, R.D.; Powell, C.A. Prevalence and Natural Host Range of Homalodisca Coagulata Virus-1 (HoCV-1). Arch. Virol. 2008, 153, 61–67. [Google Scholar] [CrossRef]
- Reinganum, C. The Isolation of Cricket Paralysis Virus from the Emperor Gum Moth, Antheraea Eucalypti Scott, and Its Infectivity towards a Range of Insect Species. Intervirology 1975, 5, 97–102. [Google Scholar] [CrossRef]
- Hough-Goldstein, J.A.; Heimpel, G.E.; Bechmann, H.E.; Mason, C.E. Arthropod Natural Enemies of the Colorado Potato Beetle. Crop Prot. 1993, 12, 324–334. [Google Scholar] [CrossRef]
- Bonning, B.C.; Nusawardani, T. Introduction to the Use of Baculoviruses as Biological Insecticides. Methods Mol. Biol. 2007, 388, 359–366. [Google Scholar] [CrossRef]
- Tanaka, H.; Sato, K.; Saito, Y.; Yamashita, T.; Agoh, M.; Okunishi, J.; Tachikawa, E.; Suzuki, K. Insect Diapause-Specific Peptide from the Leaf Beetle Has Consensus with a Putative Iridovirus Peptide. Peptides 2003, 24, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Boonham, N.; Tomlinson, J.; Mumford, R. Microarrays for Rapid Identification of Plant Viruses. Annu. Rev. Phytopathol. 2007, 45, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, L.; Rajashekara, H.; Uppala, L.S.; Ambika, D.S.; Patil, B.; Shankarappa, K.S.; Nath, V.S.; Kavitha, T.R.; Mishra, A.K. Mechanisms of Microbial Plant Protection and Control of Plant Viruses. Plants 2022, 11, 3449. [Google Scholar] [CrossRef] [PubMed]
- Petek, M.; Rotter, A.; Kogovšek, P.; Baebler, S.; Mithöfer, A.; Gruden, K. Potato Virus Y Infection Hinders Potato Defence Response and Renders Plants More Vulnerable to Colorado Potato Beetle Attack. Mol. Ecol. 2014, 23, 5378–5391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristić, D.; Vučurović, I.; Kuzmanović, S.; Pfaf-Dolovac, E.; Aleksić, G.; Vučurović, A.; Starović, M. The Incidence and Genetic Diversity of Potato Virus S in Serbian Seed Potato Crops. Potato Res. 2019, 62, 31–46. [Google Scholar] [CrossRef]
- Wickizer, S.L.; Gergerich, R.C. First Report of Japanese Beetle (Popillia japonica) as a Vector of Southern bean mosaic virus and Bean pod mottle virus. Plant Dis. 2007, 91, 637. [Google Scholar] [CrossRef]
- Wielkopolan, B.; Jakubowska, M.; Obrępalska-Stęplowska, A. Beetles as Plant Pathogen Vectors. Front. Plant Sci. 2021, 12, 748093. [Google Scholar] [CrossRef]
- Gilbert, C.; Belliardo, C. The Diversity of Endogenous Viral Elements in Insects. Curr. Opin. Insect Sci. 2022, 49, 48–55. [Google Scholar] [CrossRef]
- Palatini, U.; Contreras, C.A.; Gasmi, L.; Bonizzoni, M. Endogenous Viral Elements in Mosquito Genomes: Current Knowledge and Outstanding Questions. Curr. Opin. Insect Sci. 2022, 49, 22–30. [Google Scholar] [CrossRef]
- Palatini, U.; Miesen, P.; Carballar-Lejarazu, R.; Ometto, L.; Rizzo, E.; Tu, Z.; van Rij, R.P.; Bonizzoni, M. Comparative Genomics Shows That Viral Integrations Are Abundant and Express PiRNAs in the Arboviral Vectors Aedes aegypti and Aedes albopictus. BMC Genomics 2017, 18, 512. [Google Scholar] [CrossRef] [Green Version]
- Wallau, G.L. RNA Virus EVEs in Insect Genomes. Curr. Opin. Insect Sci. 2022, 49, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented Genomic Diversity of RNA Viruses in Arthropods Reveals the Ancestry of Negative-Sense RNA Viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, N.; Dheilly, N.M.; Junglen, S.; Paraskevopoulou, S.; Postler, T.S.; Shi, M.; Kuhn, J.H. Jingchuvirales: A New Taxonomical Framework for a Rapidly Expanding Order of Unusual Monjiviricete Viruses Broadly Distributed among Arthropod Subphyla. Appl. Environ. Microbiol. 2022, 88, e01954-21. [Google Scholar] [CrossRef]
- Lung, O.; Blissard, G.W. A Cellular Drosophila Melanogaster Protein with Similarity to Baculovirus F Envelope Fusion Proteins. J. Virol. 2005, 79, 7979–7989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrmann, G.F.; Karplus, P.A. Relatedness of Baculovirus and Gypsy Retrotransposon Envelope Proteins. BMC Evol. Biol. 2001, 1, 1. [Google Scholar] [CrossRef]
- Strand, M.R.; Drezen, J.-M. Family Polydnaviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 267–278. [Google Scholar] [CrossRef]
- Strand, M.R.; Burke, G.R. Polydnaviruses: From Discovery to Current Insights. Virology 2015, 479–480, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Gasmi, L.; Boulain, H.; Gauthier, J.; Hua-van, A.; Musset, K.; Jakubowska, A.K.; Aury, J.; Volkoff, A.; Huguet, E. Recurrent Domestication by Lepidoptera of Genes from Their Parasites Mediated by Bracoviruses. PLoS Genet. 2015, 11, e1005470. [Google Scholar] [CrossRef] [Green Version]
- Jansson, R.K.; Groden, E.; Zoology, E.; Experiment, A.; Brunswick, N. Parasitism of Leptinotarsa decemlineata [Coleoptera: Chrysomelidae] by Edovum puttleri [Hymenoptera: Eulophidae] in different cultivars of eggplant. Entomophaga 1987, 32, 503–510. [Google Scholar] [CrossRef]
- Wang, Y.; Jehle, J.A. Nudiviruses and Other Large, Double-Stranded Circular DNA Viruses of Invertebrates: New Insights on an Old Topic. J. Invertebr. Pathol. 2009, 101, 187–193. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Species | Max Alignment Length, aa | Max Identity, % | Max Contig Length, bp | Number of Contigs | Protein Product |
|---|---|---|---|---|---|---|---|
| Martellivirales | Virgaviridae | Nephila clavipes virus 4 | 1047 | 40.51 | 10,559 | 51 | RdRp |
| Broome virga-like virus 1 | 177 | 31.64 | 2499 | 5 | ORF3 | ||
| Insect virga-like virus 1 | 165 | 41.67 | 540 | 2 | Hypothetical protein | ||
| Tymovirales | Betaflexiviridae | Potato virus S | 1975 | 100 | 8539 | 66 | Structural and nonstructural polyprotein |
| Bunyavirales | Phasmaviridae | Wuhan mosquito orthophasmavirus 1 | 388 | 27.06 | 1667 | 17 | Glycoprotein |
| Articulavirales | Orthomyxoviridae | Quaranjavirus araguariense | 437 | 27.82 | 1718 | 9 | Hemagglutinin |
| Photinus pyralis orthomyxo-like virus 1 | 768 | 43.94 | 2529 | 15 | Nucleocapsid protein, polymerase PB2 | ||
| Coleopteran orthomyxo-related virus OKIAV184 | 501 | 31.34 | 1715 | 2 | Hemagglutinin | ||
| Coleopteran orthomyxo-related virus OKIAV186 | 761 | 46.78 | 2607 | 2 | Polymerase PB2 | ||
| Jingchuvirales | Chuviridae | Nigecruvirus ixodes | 631 | 36.42 | 2176 | 14 | Glycoprotein |
| Coleopteran chu-related virus OKIAV127 | 167 | 72.79 | 1926 | 22 | |||
| Orthopteran chu-related virus OKIAV152 | 138 | 38.41 | 2075 | 1 | |||
| Mononegavirales | Xinmoviridae | Orthopteran anphevirus | 383 | 25.10 | 1645 | 3 | Hypothetical protein 1 |
| Ulegvirus freckenfeldense | 1930 | 30.95 | 14,588 | 9 | RdRp | ||
| Pimascovirales | Ascoviridae | Trichoplusia ni ascovirus 2c | 137 | 40.63 | 1321 | 19 | Hypothetical protein |
| Pimascovirales | Iridoviridae | Iridovirus LCIVAC01 | 168 | 30.95 | 796 | 2 | Uncharacterized protein |
| Picornavirales | Caliciviridae | Soybean thrips Pernambuco virus | 497 | 25.81 | 11,259 | 6 | Polyprotein |
| Dicistroviridae | Aphid lethal paralysis virus | 808 | 92.82 | 3468 | 1 | Capsid protein | |
| Aphis gossypii virus | 1039 | 93.94 | 6635 | 1 | ORF1 | ||
| Bivalve RNA virus G5 | 369 | 24.93 | 6436 | 1 | Replicative protein | ||
| Iflaviridae | Lampyris noctiluca iflavirus 1 | 1642 | 39.73 | 9934 | 26 | Putative polyprotein | |
| Solinviviridae | Solenopsis invicta virus 3 | 966 | 29.12 | 11,648 | 12 | Structural and nonstructural polyprotein | |
| Patatavirales | Potyviridae | Potato virus Y | 3061 | 100 | 9728 | 25 | Coat protein, polyprotein |
| Lefavirales | Baculoviridae | Mythimna unipuncta nucleopolyhedrovirus | 309 | 25.21 | 1929 | 14 | F-protein |
| Peridroma alphabaculovirus | 474 | 30.95 | 2668 | 9 | |||
| Mythimna unipuncta granulovirus B | 251 | 25.10 | 1628 | 14 | |||
| Helicoverpa armigera nucleopolyhedrovirus | 276 | 22.60 | 1550 | 4 | Envelope protein, hypothetical protein | ||
| Lambdina fiscellaria nucleopolyhedrovirus | 156 | 35.90 | 962 | 1 | Vp80 | ||
| Plutella xylostella granulovirus | 271 | 21.03 | 1275 | 1 | PxORF26 peptide | ||
| Xestia c-nigrum granulovirus | 220 | 26.82 | 1394 | 1 | ORF27 | ||
| Nudiviridae | Penaeus monodon nudivirus | 655 | 28.55 | 17,638 | 1 | P74 | |
| Polydnaviriformidae | Bracoviriform congregatae | 242 | 71.43 | 1669 | 14 | Structural and nonstructural protein | |
| Bracoviriform inaniti | 419 | 43.46 | 2975 | 28 | |||
| Cotesia sesamiae bracovirus | 614 | 54.76 | 6071 | 19 | |||
| Bracoviriform facetosae | 158 | 25.95 | 716 | 1 | |||
| Wuhan insect virus 33 | 1039 | 99.32 | 6635 | 1 | Hypothetical protein | ||
| Papaya mottle associated virus | 1017 | 64.40 | 8539 | 63 | RdRp | ||
| Old quarry swamp virus | 794 | 46.36 | 2607 | 20 | Putative nucleocapsid, polymerase | ||
| Allermuir Hill virus 1 | 1016 | 46.63 | 9840 | 17 | Putative nonstructural polyprotein |
| Family | Species | Coverage, Bases | Coverage, % | Genome Length | Projects | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Median | Min | Max | Median | Min | Max | |||
| Potyviridae | Potato virus Y | 55 | 9561 | 100.50 | 0.57 | 98.63 | 1.03 | 9595 | 9800 | 8 |
| Betaflexiviridae | Potato virus S | 55 | 8481 | 6336.00 | 0.65 | 99.95 | 74.56 | 8453 | 8515 | 10 |
| Dicistroviridae | Aphis glycines virus 3 | 133 | 6691 | 3412.00 | 1.31 | 70.84 | 36.08 | 9445 | 10,131 | 2 |
| Aphid lethal paralysis virus | 6635 | 6635 | 6635.00 | 67.57 | 67.57 | 67.57 | 9819 | 9819 | 1 | |
| Betaflexiviridae | Potato virus H | 6520 | 6520 | 6520.00 | 77.53 | 77.53 | 77.53 | 8410 | 8410 | 1 |
| Unclassified | Wuhan insect virus 33 | 5877 | 5877 | 5877.00 | 57.44 | 57.44 | 57.44 | 10,232 | 10,232 | 1 |
| Dicistroviridae | Homalodisca coagulata virus 1 | 3998 | 3998 | 3998.00 | 42.78 | 42.78 | 42.78 | 9345 | 9345 | 1 |
| Retroviridae | Human endogenous retrovirus K | 90 | 2908 | 277.00 | 0.95 | 30.70 | 2.92 | 9472 | 9472 | 6 |
| Virgaviridae | Turnip vein-clearing virus | 2795 | 2795 | 2795.00 | 44.29 | 44.29 | 44,29 | 6311 | 6311 | 1 |
| Herpesviridae | Human betaherpesvirus 5 | 50 | 2647 | 191.00 | 0.02 | 1.15 | 0.08 | 229,209 | 235,732 | 14 |
| Togaviridae | Semliki Forest virus | 100 | 1838 | 622.00 | 0.69 | 12.65 | 4.28 | 14,529 | 14,529 | 12 |
| Unclassified | Aphis glycines virus 2 | 1567 | 1567 | 1567.00 | 32.31 | 32.31 | 32.31 | 4850 | 4850 | 1 |
| Hubei picorna-like virus 15 | 1395 | 1395 | 1395.00 | 14.08 | 14.08 | 14.08 | 9910 | 9910 | 1 | |
| Dicistroviridae | Rhopalosiphum padi virus | 1235 | 1235 | 1235.00 | 12.29 | 12.29 | 12.29 | 10,045 | 10,045 | 1 |
| Unclassified | Wuhan aphid virus 1 | 1185 | 1185 | 1185.00 | 41.89 | 41.89 | 41.89 | 2829 | 2829 | 1 |
| Baculoviridae | Autographa californica multiple nucleopolyhedrovirus | 50 | 1086 | 294.00 | 0.04 | 0.92 | 0.23 | 118,582 | 138,991 | 8 |
| Dicistroviridae | Big Sioux River virus | 1064 | 1064 | 1064.00 | 10.39 | 10.39 | 10.39 | 10,237 | 10,237 | 1 |
| Paramyxoviridae | Canine morbillivirus | 105 | 1017 | 453.00 | 0.56 | 5.40 | 2.41 | 18,826 | 18,826 | 12 |
| Adenoviridae | Human mastadenovirus C | 55 | 775 | 182.00 | 0.15 | 2.16 | 0.51 | 35,926 | 35,958 | 5 |
| Polydnaviriformidae | Chelonus inanitus bracovirus | 368 | 722 | 545.00 | 1.72 | 3.38 | 2.55 | 21,376 | 21,376 | 2 |
| Flaviviridae | Pestivirus A | 36 | 401 | 92.50 | 0.25 | 3.16 | 0.68 | 12,295 | 15,752 | 14 |
| Pospiviroidae | Citrus exocortis viroid | 121 | 376 | 214.00 | 30.56 | 94.95 | 54.04 | 396 | 396 | 13 |
| Polydnaviriformidae | Diolcogaster facetosa bracovirus | 73 | 345 | 101.00 | 0.07 | 0.98 | 0.10 | 12,859 | 104,039 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starchevskaya, M.; Kamanova, E.; Vyatkin, Y.; Tregubchak, T.; Bauer, T.; Bodnev, S.; Rotskaya, U.; Polenogova, O.; Kryukov, V.; Antonets, D. The Metagenomic Analysis of Viral Diversity in Colorado Potato Beetle Public NGS Data. Viruses 2023, 15, 395. https://doi.org/10.3390/v15020395
Starchevskaya M, Kamanova E, Vyatkin Y, Tregubchak T, Bauer T, Bodnev S, Rotskaya U, Polenogova O, Kryukov V, Antonets D. The Metagenomic Analysis of Viral Diversity in Colorado Potato Beetle Public NGS Data. Viruses. 2023; 15(2):395. https://doi.org/10.3390/v15020395
Chicago/Turabian StyleStarchevskaya, Maria, Ekaterina Kamanova, Yuri Vyatkin, Tatyana Tregubchak, Tatyana Bauer, Sergei Bodnev, Ulyana Rotskaya, Olga Polenogova, Vadim Kryukov, and Denis Antonets. 2023. "The Metagenomic Analysis of Viral Diversity in Colorado Potato Beetle Public NGS Data" Viruses 15, no. 2: 395. https://doi.org/10.3390/v15020395