
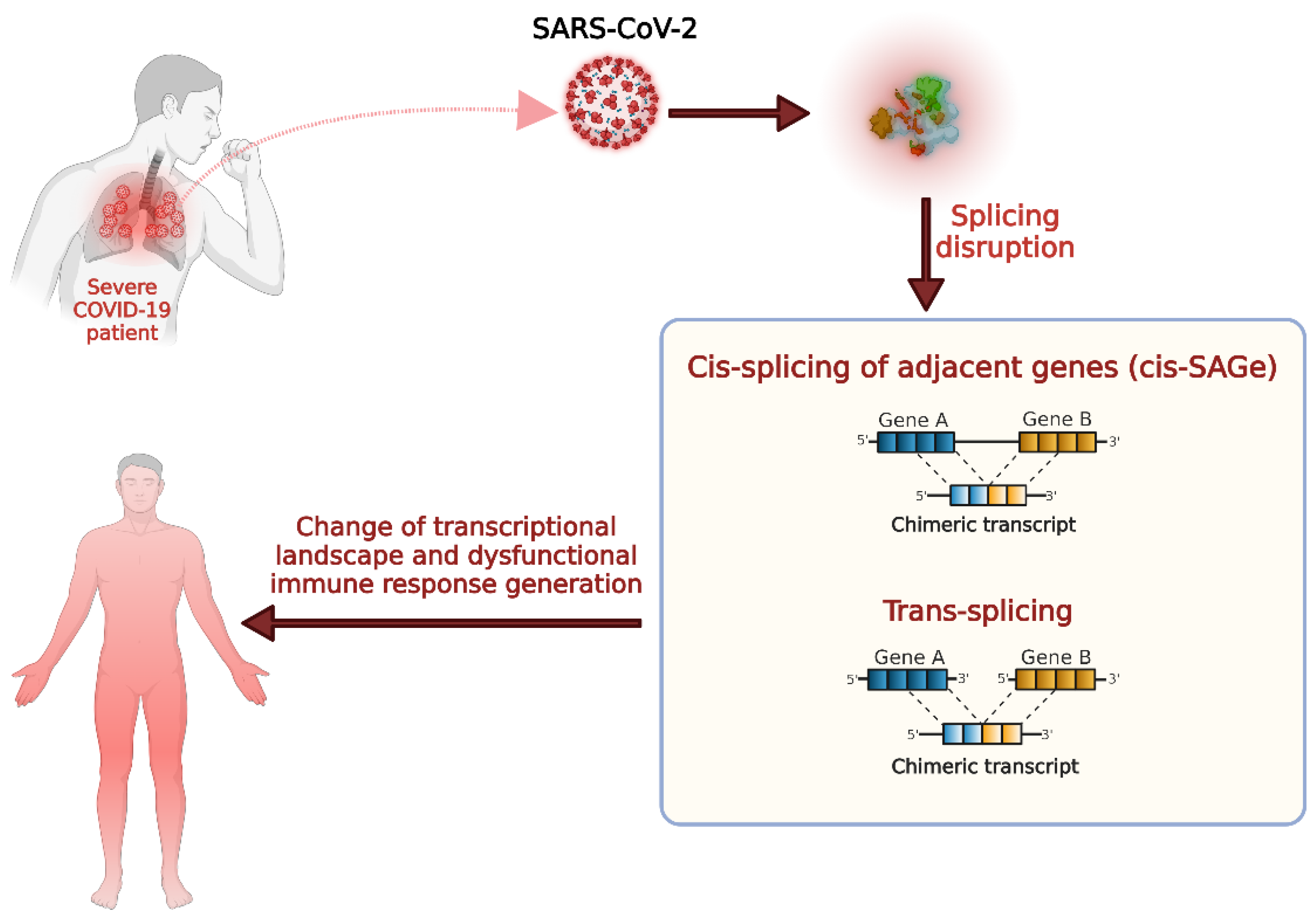
The Landscape of Expressed Chimeric Transcripts in the Blood of Severe COVID-19 Infected Patients

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of the RNA-Seq Datasets
2.2. Identification of Chimeric Transcripts from the RNA-Seq Data
2.3. Differential Gene Expression Analysis
2.4. Gene Ontology and Pathway Enrichment Analysis of the Parental Genes of Chimeric Transcripts
3. Results
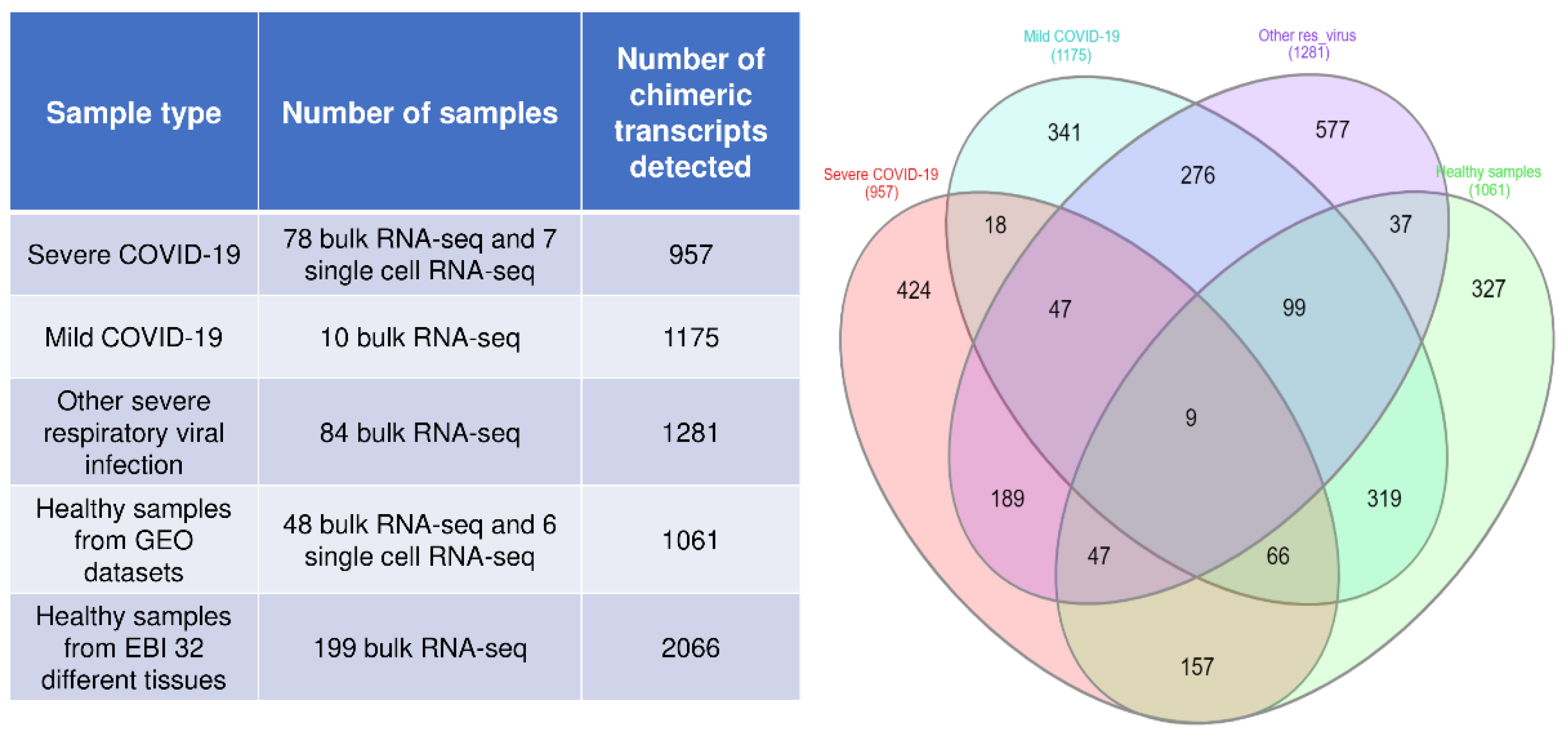
3.1. Identification of Chimeric Transcripts in the RNA-Seq Data of Blood Samples from Severe COVID-19, Mild COVID-19, and Other Severe Respiratory Virus-Infected Patients
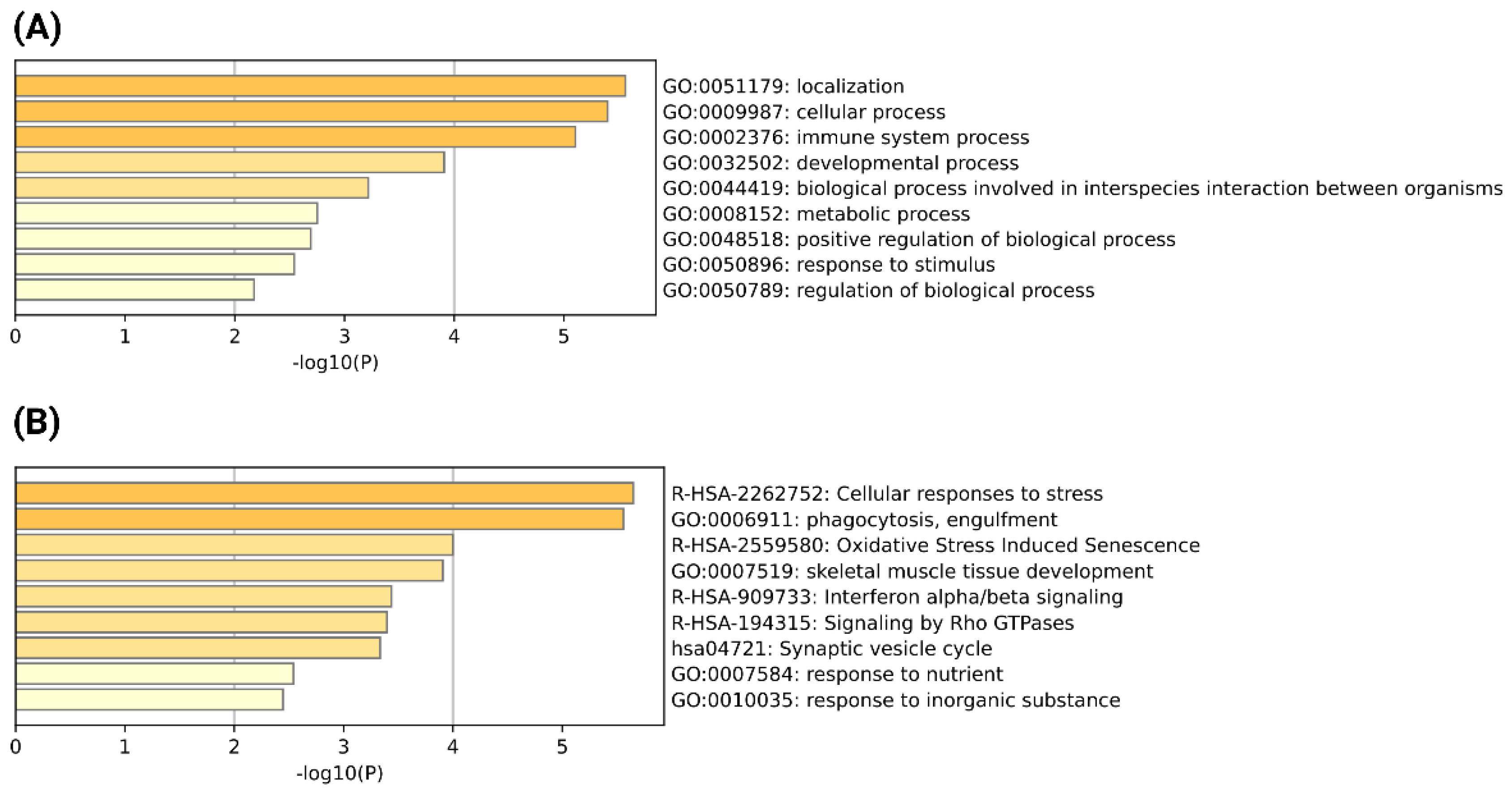
3.2. Identification of Severe COVID-19 Specific Recurrent Chimeric Transcripts and Functional Analysis of Their Parental Genes
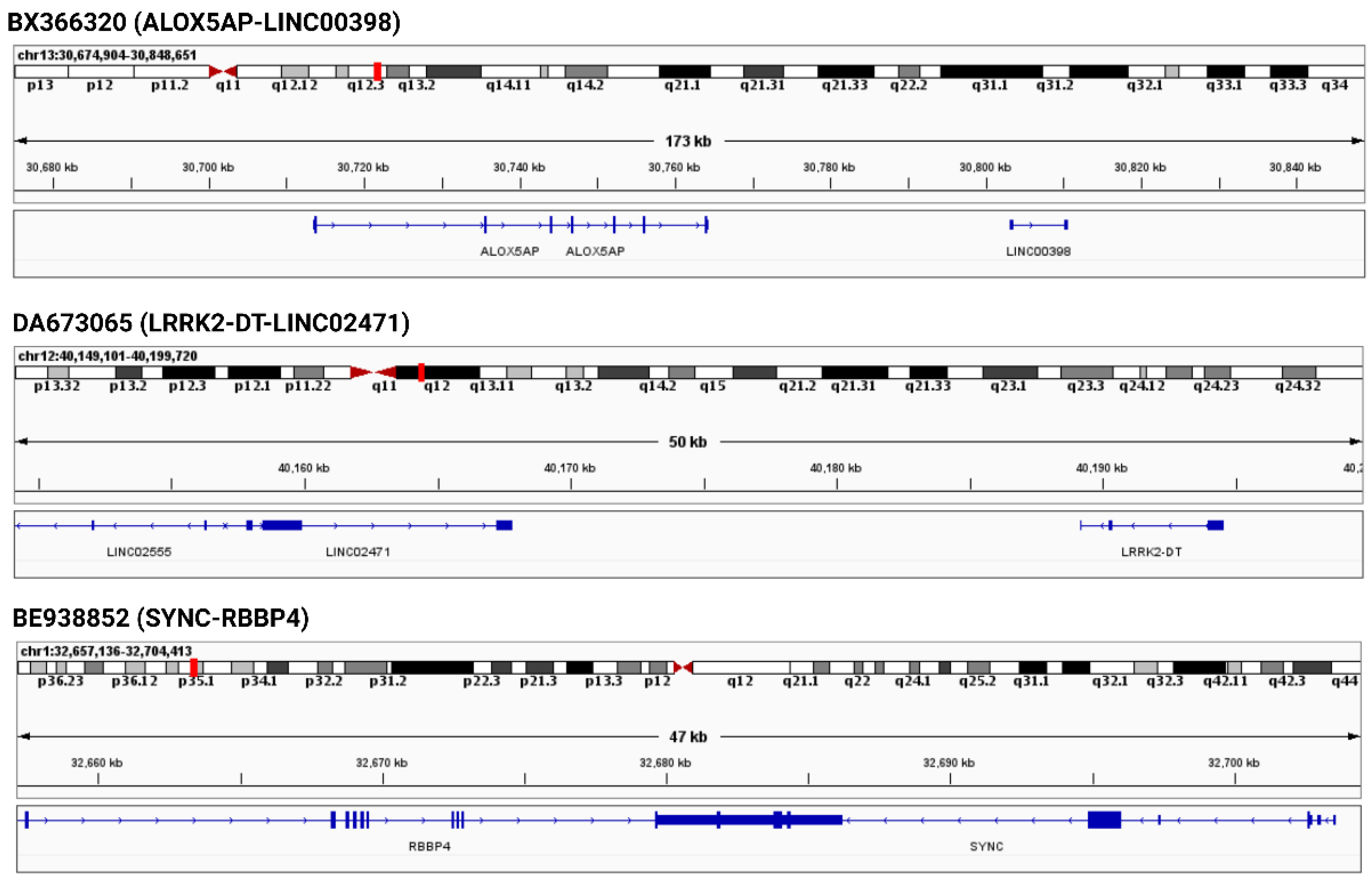
3.3. Genomic Neighborhood Analysis and Differential Gene Expression Analysis of the Parental Genes of Severe COVID-19 Specific Recurrent Chimeric Transcripts
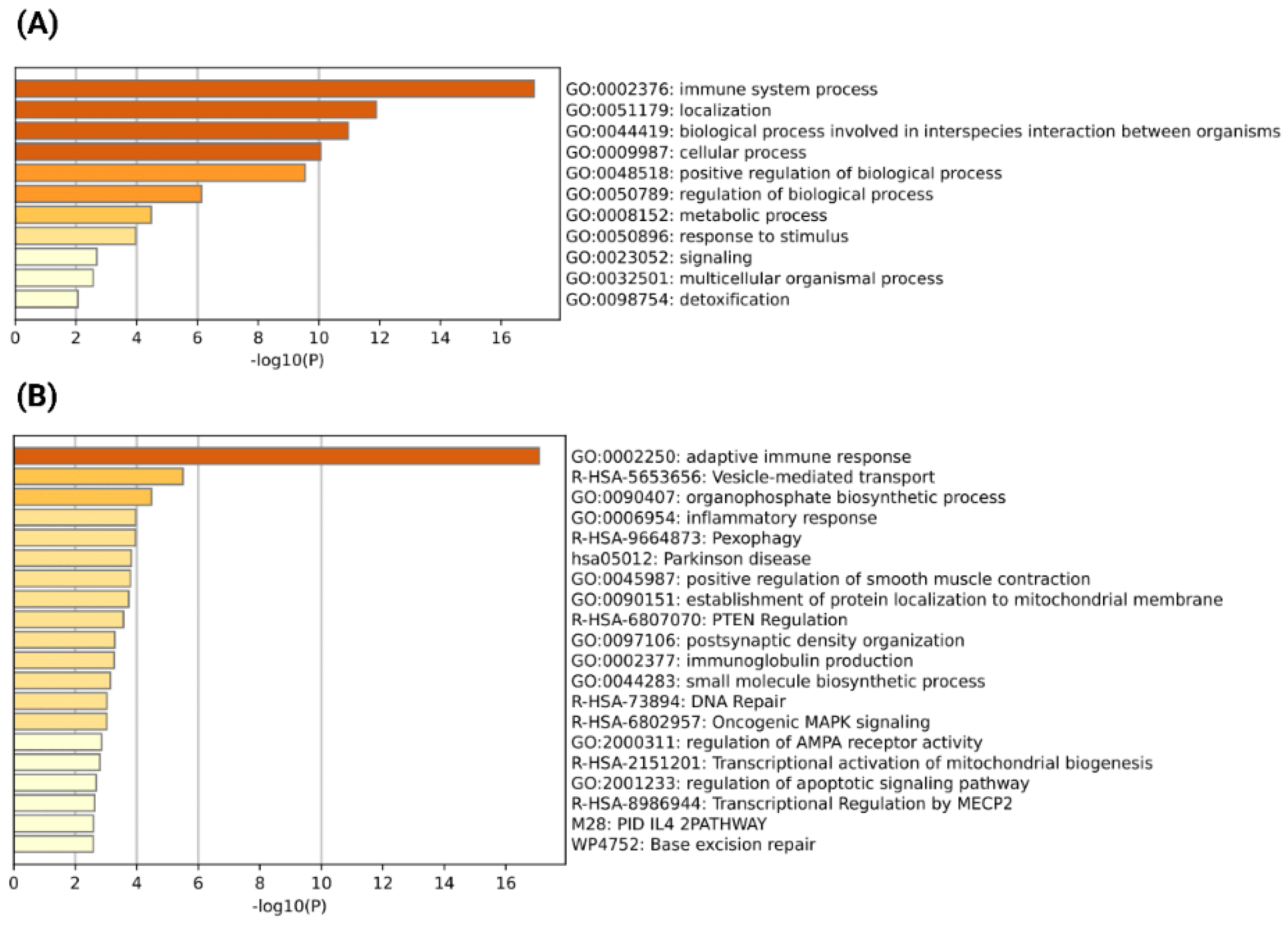
3.4. Identification of Common Chimeric Transcripts Expressed in Severe COVID-19 and Other Severe Respiratory Viral Infections
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus Correctassociated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Fan, E.; Beitler, J.R.; Brochard, L.; Calfee, C.S.; Ferguson, N.D.; Slutsky, A.S.; Brodie, D. COVID-19-associated acute respiratory distress syndrome: Is a different approach to management warranted? Lancet Respir. Med. 2020, 8, 816–821. [Google Scholar] [CrossRef]
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe COVID-19. N. Engl. J. Med. 2020, 383, 2451–2460. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Simbaña-Rivera, K.; Gómez- Barreno, L.; Rubio-Neira, M.; Guaman, L.P.; Kyriakidis, N.C.; Muslin, C.; Jaramillo, A.M.G.; Barba-Ostria, C.; Cevallos-Robalino, D.; et al. Clinical, molecular, and epidemiological characterization of the SARS-CoV-2 virus and the Coronavirus Disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115094. [Google Scholar] [CrossRef]
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Munnink, B.B.O.; De Meulder, D.; Van Amerongen, G.; Van Den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020, 368, 1012–1015. [Google Scholar] [CrossRef]
- Mukherjee, S.; Banerjee, B.; Karasik, D.; Frenkel-Morgenstern, M. mRNA-lncRNA Co-Expression Network Analysis Reveals the Role of lncRNAs in Immune Dysfunction during Severe SARS-CoV-2 Infection. Viruses 2021, 13, 402. [Google Scholar] [CrossRef]
- Vaninov, N. In the eye of the COVID-19 cytokine storm. Nat. Rev. Immunol. 2020, 20, 277. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Wang, C.; Chen, L.; Chen, Y.; Jia, W.; Cai, X.; Liu, Y.; Ji, F.; Xiong, P.; Liang, A.; Liu, R.; et al. Abnormal global alternative RNA splicing in COVID-19 patients. PLoS Genet. 2022, 18, e1010137. [Google Scholar] [CrossRef]
- Mukherjee, S.B.; Mukherjee, S.; Detroja, R.; Frenkel-Morgenstern, M. The landscape of differential splicing and transcript alternations in severe COVID-19 infection. FEBS J. 2023. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; Expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Zhuo, J.-S.; Jing, X.-Y.; Du, X.; Yang, X.-Q. Generation of Chimeric RNAs by cis-splicing of adjacent genes (cis-SAGe) in mammals. Yi Chuan Hered. 2018, 40, 145–154. [Google Scholar]
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef]
- Shi, X.; Singh, S.; Lin, E.; Li, H. Chimeric RNAs in cancer. In Advances in Clinical Chemistry; Academic Press Inc.: Cambridge, MA, USA, 2021; Volume 100, pp. 1–35. ISBN 9780128239223. [Google Scholar]
- Pitolli, C.; Marini, A.; Sette, C.; Pagliarini, V. Non-Canonical Splicing and Its Implications in Brain Physiology and Cancer. Int. J. Mol. Sci. 2022, 23, 2811. [Google Scholar] [CrossRef]
- Chwalenia, K.; Facemire, L.; Li, H. Chimeric RNAs in cancer and normal physiology. Wiley Interdiscip. Rev. RNA 2017, 8, e1427. [Google Scholar] [CrossRef]
- Mukherjee, S.; Heng, H.H.; Frenkel-Morgenstern, M. Emerging Role of Chimeric RNAs in Cell Plasticity and Adaptive Evolution of Cancer Cells. Cancers 2021, 13, 4328. [Google Scholar] [CrossRef]
- Detroja, R.; Mukherjee, S.; Frenkel-Morgenstern, M. The Landscape of Novel Expressed Chimeric RNAs in Rheumatoid Arthritis. Cells 2022, 11, 1092. [Google Scholar] [CrossRef]
- Mukherjee, S.; Frenkel-Morgenstern, M. Evolutionary impact of chimeric RNAs on generating phenotypic plasticity in human cells. Trends Genet. 2022, 38, 4–7. [Google Scholar] [CrossRef]
- Mukherjee, S.; Mukherjee, S.B.; Frenkel-Morgenstern, M. Functional and regulatory impact of chimeric RNAs in human normal and cancer cells. Wiley Interdiscip. Rev. RNA 2023, e1777. [Google Scholar] [CrossRef]
- Mukherjee, S.B.; Mukherjee, S.; Frenkel-Morgenstern, M. Fusion proteins mediate alternation of protein interaction networks in cancers. In Advances in Protein Chemistry and Structural Biology; Academic Press Inc.: Cambridge, MA, USA, 2022; Volume 131, pp. 165–176. ISBN 9780323992312. [Google Scholar]
- Frenkel-Morgenstern, M.; Lacroix, V.; Ezkurdia, I.; Levin, Y.; Gabashvili, A.; Prilusky, J.; Del Pozo, A.; Tress, M.; Johnson, R.; Guigo, R.; et al. Chimeras taking shape: Potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012, 22, 1231–1242. [Google Scholar] [CrossRef]
- Latysheva, N.S.; Babu, M.M. Molecular Signatures of Fusion Proteins in Cancer. ACS Pharmacol. Transl. Sci. 2019, 2, 122–133. [Google Scholar] [CrossRef]
- Lévy, Y.; Wiedemann, A.; Hejblum, B.P.; Durand, M.; Lefebvre, C.; Surénaud, M.; Lacabaratz, C.; Perreau, M.; Foucat, E.; Déchenaud, M.; et al. CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience 2021, 24, 102711. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.P.M.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tsang, O.T.Y.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Jackson, H.; Rivero Calle, I.; Broderick, C.; Habgood-Coote, D.; D’Souza, G.; Nichols, S.; Vito, O.; Gómez-Rial, J.; Rivero-Velasco, C.; Rodríguez-Núñez, N.; et al. Characterisation of the blood RNA host response underpinning severity in COVID-19 patients. Sci. Rep. 2022, 12, 12216. [Google Scholar] [CrossRef] [PubMed]
- Tsalik, E.L.; Fiorino, C.; Aqeel, A.; Liu, Y.; Henao, R.; Ko, E.R.; Burke, T.W.; Reller, M.E.; Bodinayake, C.K.; Nagahawatte, A.; et al. The Host Response to Viral Infections Reveals Common and Virus-Specific Signatures in the Peripheral Blood. Front. Immunol. 2021, 12, 741837. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Detroja, R.; Gorohovski, A.; Giwa, O.; Baum, G.; Frenkel-Morgenstern, M. ChiTaH: A fast and accurate tool for identifying known human chimeric sequences from high-throughput sequencing data. NAR Genom. Bioinform. 2021, 3, lqab112. [Google Scholar] [CrossRef]
- Benelli, M.; Pescucci, C.; Marseglia, G.; Severgnini, M.; Torricelli, F.; Magi, A. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 2012, 28, 3232–3239. [Google Scholar] [CrossRef]
- Haas, B.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.; Pochet, N. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv 2017. [Google Scholar] [CrossRef]
- Davidson, N.M.; Majewski, I.J.; Oshlack, A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015, 7, 43. [Google Scholar] [CrossRef]
- Nicorici, D.; Satalan, M.; Edgren, H.; Kangaspeska, S.; Murumagi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014. [Google Scholar] [CrossRef]
- Balamurali, D.; Gorohovski, A.; Detroja, R.; Palande, V.; Raviv-Shay, D.; Frenkel-Morgenstern, M. ChiTaRS 5.0: The comprehensive database of chimeric transcripts matched with druggable fusions and 3D chromatin maps. Nucleic Acids Res. 2019, 48, D825–D834. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Anders, S.; Huber, W. Differential analysis of count data—The DESeq2 package. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, W. Benjamini–Hochberg Method. In Encyclopedia of Systems Biology; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 48, D498–D503. [Google Scholar] [CrossRef]
- Mukherjee, S.; Detroja, R.; Balamurali, D.; Matveishina, E.; Medvedeva, Y.A.; Valencia, A.; Gorohovski, A.; Frenkel-Morgenstern, M. Computational analysis of sense-antisense chimeric transcripts reveals their potential regulatory features and the landscape of expression in human cells. NAR Genom. Bioinform. 2021, 3, lqab074. [Google Scholar] [CrossRef]
- Frenkel-Morgenstern, M.; Gorohovski, A.; Tagore, S.; Sekar, V.; Vazquez, M.; Valencia, A. ChiPPI: A novel method for mapping chimeric protein-protein interactions uncovers selection principles of protein fusion events in cancer. Nucleic Acids Res. 2017, 45, 7094–7105. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Zhang, Y.; Liu, J.; Li, H. SLC45A3-ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 2017, 404, 53–61. [Google Scholar] [CrossRef]
- Guo, M.; Xiao, Z.D.; Dai, Z.; Zhu, L.; Lei, H.; Diao, L.T.; Xiong, Y. The landscape of long noncoding RNA-involved and tumor-specific fusions across various cancers. Nucleic Acids Res. 2020, 48, 12618–12631. [Google Scholar] [CrossRef]
- Sun, Y.; Li, H. Chimeric RNAs Discovered by RNA Sequencing and Their Roles in Cancer and Rare Genetic Diseases. Genes 2022, 13, 741. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, Q.; Li, F.; Zhao, W.; Xu, H.; Zhang, W.; Deng, H.; Yang, X. Identification of the cross-strand chimeric RNAs generated by fusions of bi-directional transcripts. Nat. Commun. 2021, 12, 4645. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Jividen, K.; Li, H. Chimeric RNAs generated by intergenic splicing in normal and cancer cells. Genes Chromosom. Cancer 2014, 53, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Song, Z.; Babiceanu, M.; Song, Y.; Facemire, L.; Singh, R.; Adli, M.; Li, H. Discovery of CTCF-Sensitive Cis-Spliced Fusion RNAs between Adjacent Genes in Human Prostate Cells. PLoS Genet. 2015, 11, e1005001. [Google Scholar] [CrossRef]
- Qin, F.; Song, Y.; Zhang, Y.; Facemire, L.; Frierson, H.; Li, H. Role of CTCF in regulating SLC45A3-ELK4 chimeric RNA. PLoS One 2016, 11, e0150382. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Song, Z.; Chang, M.; Song, Y.; Frierson, H.; Li, H. Recurrent cis-SAGe chimeric RNA, D2HGDH-GAL3ST2, in prostate cancer. Cancer Lett. 2016, 380, 39–46. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, M.; Yuan, H.; Park, H.G.; Frierson, H.F.; Li, H. Chimeric transcript generated by cis- splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012, 2, 598–607. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Ripa, I.; López-Guerrero, J.A. Extracellular vesicles in viral spread and antiviral response. Viruses 2020, 12, 623. [Google Scholar] [CrossRef]
- Meckes, D.G.; Raab-Traub, N. Microvesicles and Viral Infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef]
- van Dongen, H.M.; Masoumi, N.; Witwer, K.W.; Pegtel, D.M. Extracellular Vesicles Exploit Viral Entry Routes for Cargo Delivery. Microbiol. Mol. Biol. Rev. 2016, 80, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.; Mandrekar, P. Cellular stress response and innate immune signaling: Integrating pathways in host defense and inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvedaraite, E.; Hertwig, L.; Sinha, I.; Ponzetta, A.; Myrberg, I.H.; Lourda, M.; Dzidic, M.; Akber, M.; Klingström, J.; Folkesson, E.; et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl. Acad. Sci. USA 2021, 118, e2018587118. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Koenis, D.S.; Beegun, I.; Jouvene, C.C.; Aguirre, G.A.; Souza, P.R.; Gonzalez-Nunez, M.; Ly, L.; Pistorius, K.; Kocher, H.M.; Ricketts, W.; et al. Disrupted Resolution Mechanisms Favor Altered Phagocyte Responses in COVID-19. Circ. Res. 2021, 129, e54–e71. [Google Scholar] [CrossRef]
- Eskandarian Boroujeni, M.; Sekrecka, A.; Antonczyk, A.; Hassani, S.; Sekrecki, M.; Nowicka, H.; Lopacinska, N.; Olya, A.; Kluzek, K.; Wesoly, J.; et al. Dysregulated Interferon Response and Immune Hyperactivation in Severe COVID-19: Targeting STATs as a Novel Therapeutic Strategy. Front. Immunol. 2022, 13, 888897. [Google Scholar] [CrossRef] [PubMed]
- Soltani-Zangbar, M.S.; Parhizkar, F.; Ghaedi, E.; Tarbiat, A.; Motavalli, R.; Alizadegan, A.; Aghebati-Maleki, L.; Rostamzadeh, D.; Yousefzadeh, Y.; Jadideslam, G.; et al. A comprehensive evaluation of the immune system response and type-I Interferon signaling pathway in hospitalized COVID-19 patients. Cell Commun. Signal. 2022, 20, 106. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.B.; Detroja, R.; Mukherjee, S.; Frenkel-Morgenstern, M. The Landscape of Expressed Chimeric Transcripts in the Blood of Severe COVID-19 Infected Patients. Viruses 2023, 15, 433. https://doi.org/10.3390/v15020433
Mukherjee SB, Detroja R, Mukherjee S, Frenkel-Morgenstern M. The Landscape of Expressed Chimeric Transcripts in the Blood of Severe COVID-19 Infected Patients. Viruses. 2023; 15(2):433. https://doi.org/10.3390/v15020433
Chicago/Turabian StyleMukherjee, Sunanda Biswas, Rajesh Detroja, Sumit Mukherjee, and Milana Frenkel-Morgenstern. 2023. "The Landscape of Expressed Chimeric Transcripts in the Blood of Severe COVID-19 Infected Patients" Viruses 15, no. 2: 433. https://doi.org/10.3390/v15020433
APA StyleMukherjee, S. B., Detroja, R., Mukherjee, S., & Frenkel-Morgenstern, M. (2023). The Landscape of Expressed Chimeric Transcripts in the Blood of Severe COVID-19 Infected Patients. Viruses, 15(2), 433. https://doi.org/10.3390/v15020433