
Expansion of Kuravirus-like Phage Sequences within the Past Decade, including Escherichia Phage YF01 from Japan, Prompt the Creation of Three New Genera

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Media
2.2. Isolation and Purification of Phages
2.3. Transmission Electron Microscopy
2.4. Verification of pH and Temperature Stability
2.5. Phage DNA Isolation
2.6. Phage DNA Sequencing and Assembly
2.7. Phylogenetic Analysis
2.8. Genome Annotation
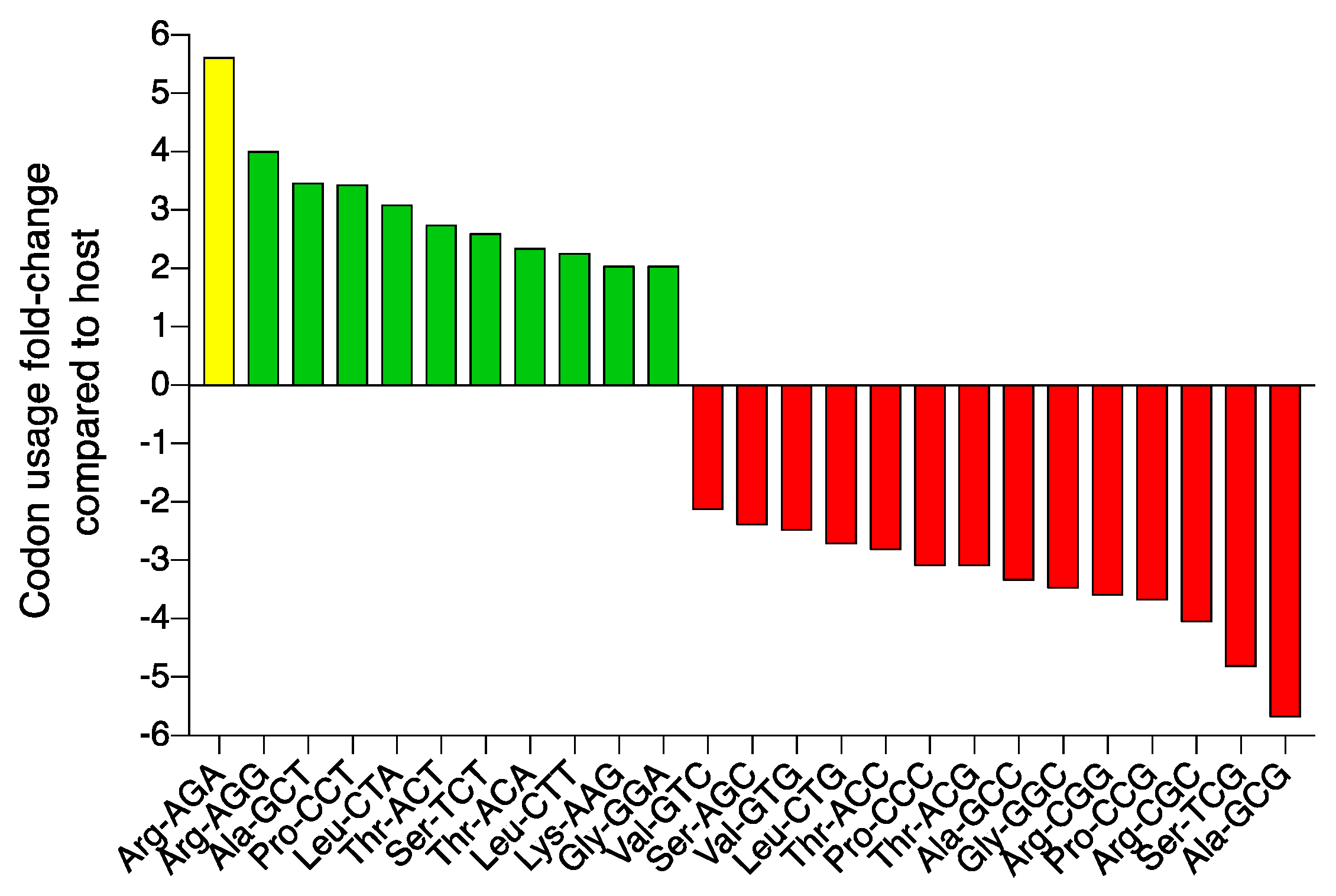
2.9. Codon Usage Analysis
2.10. Pangenome Analysis
3. Results and Discussion
3.1. Isolation and Characteristics of Escherichia Phage YF01
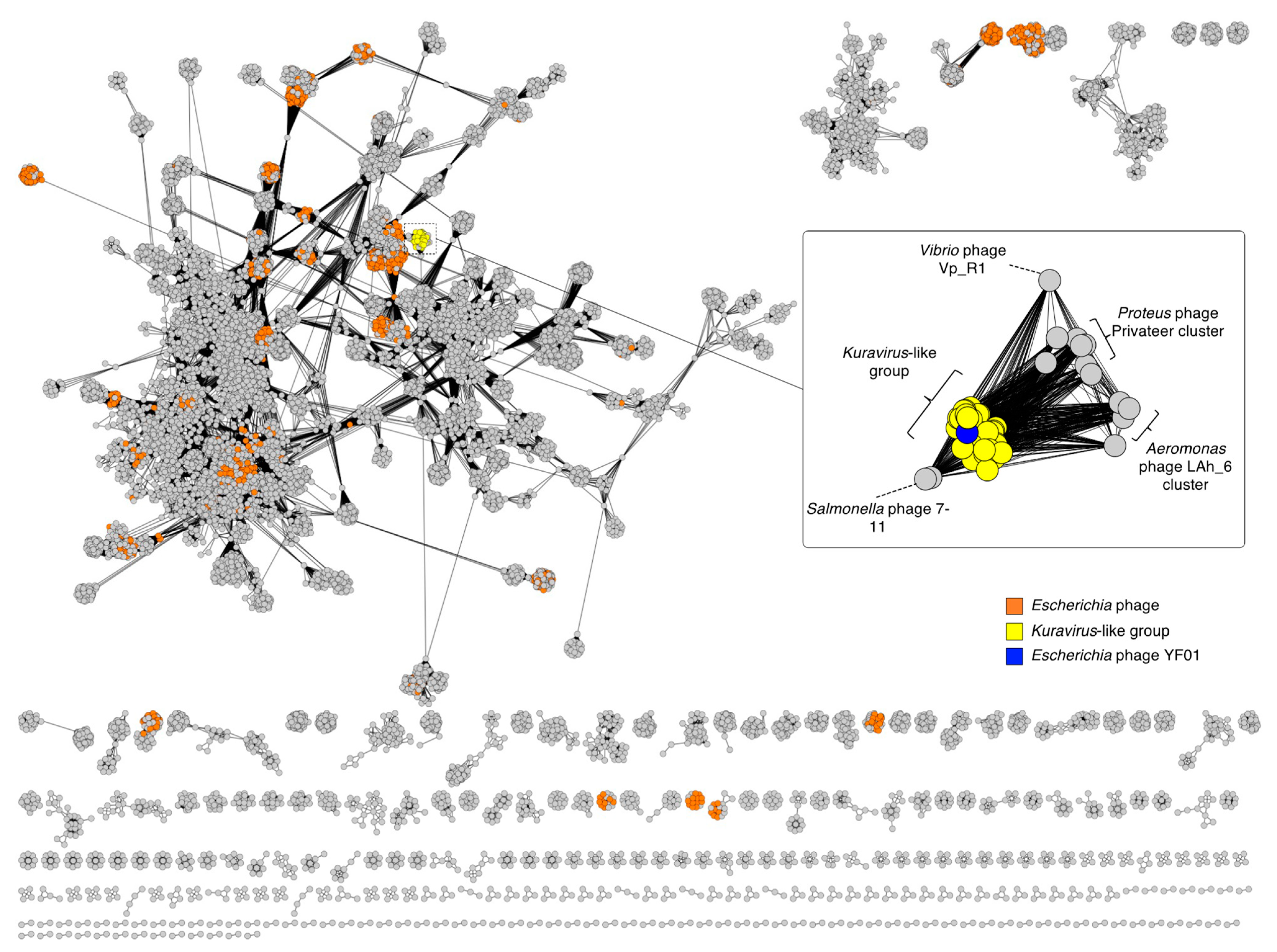
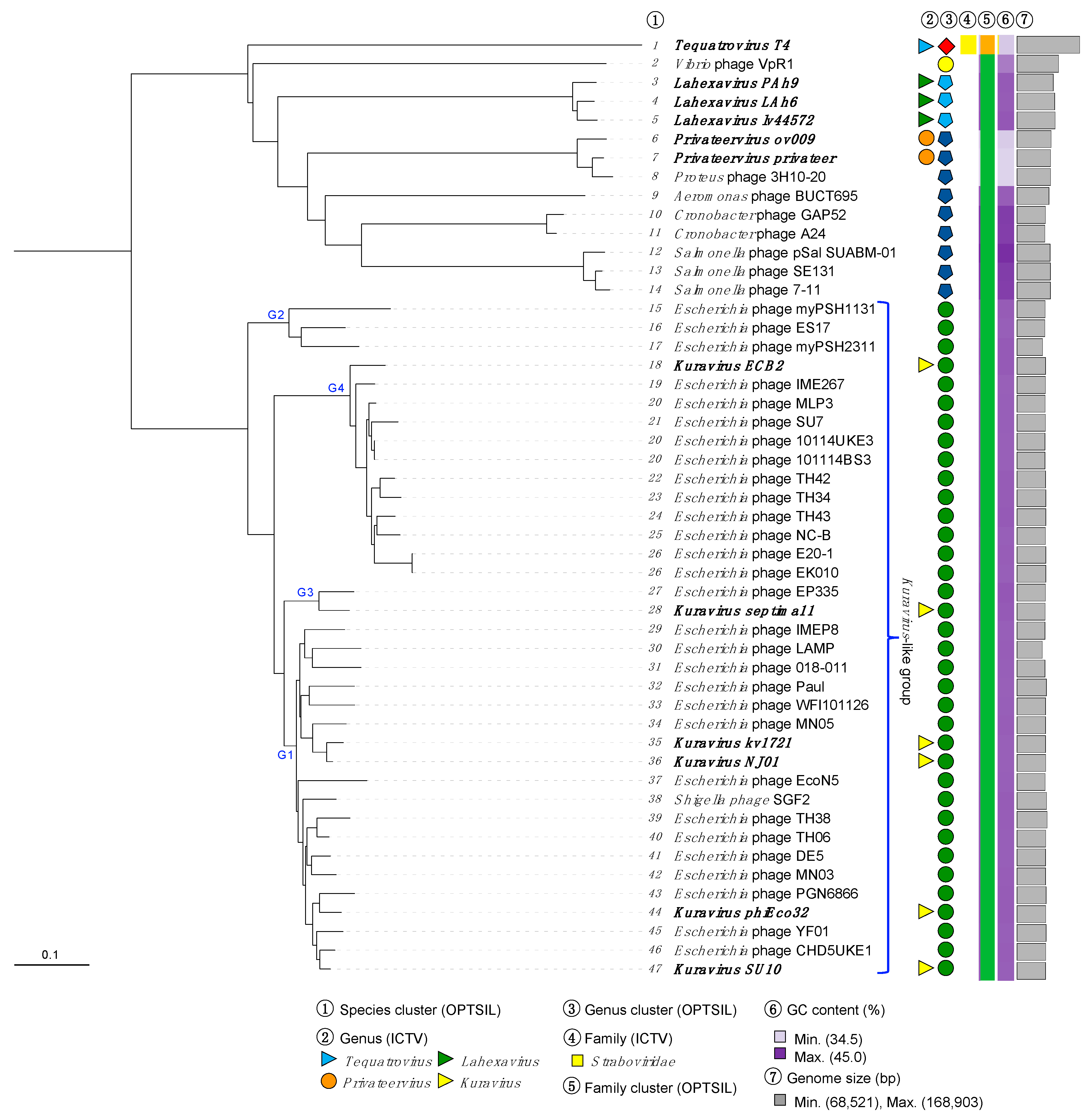
3.2. Genome Assembly and Phylogenetic Placement of the YF01 Phage
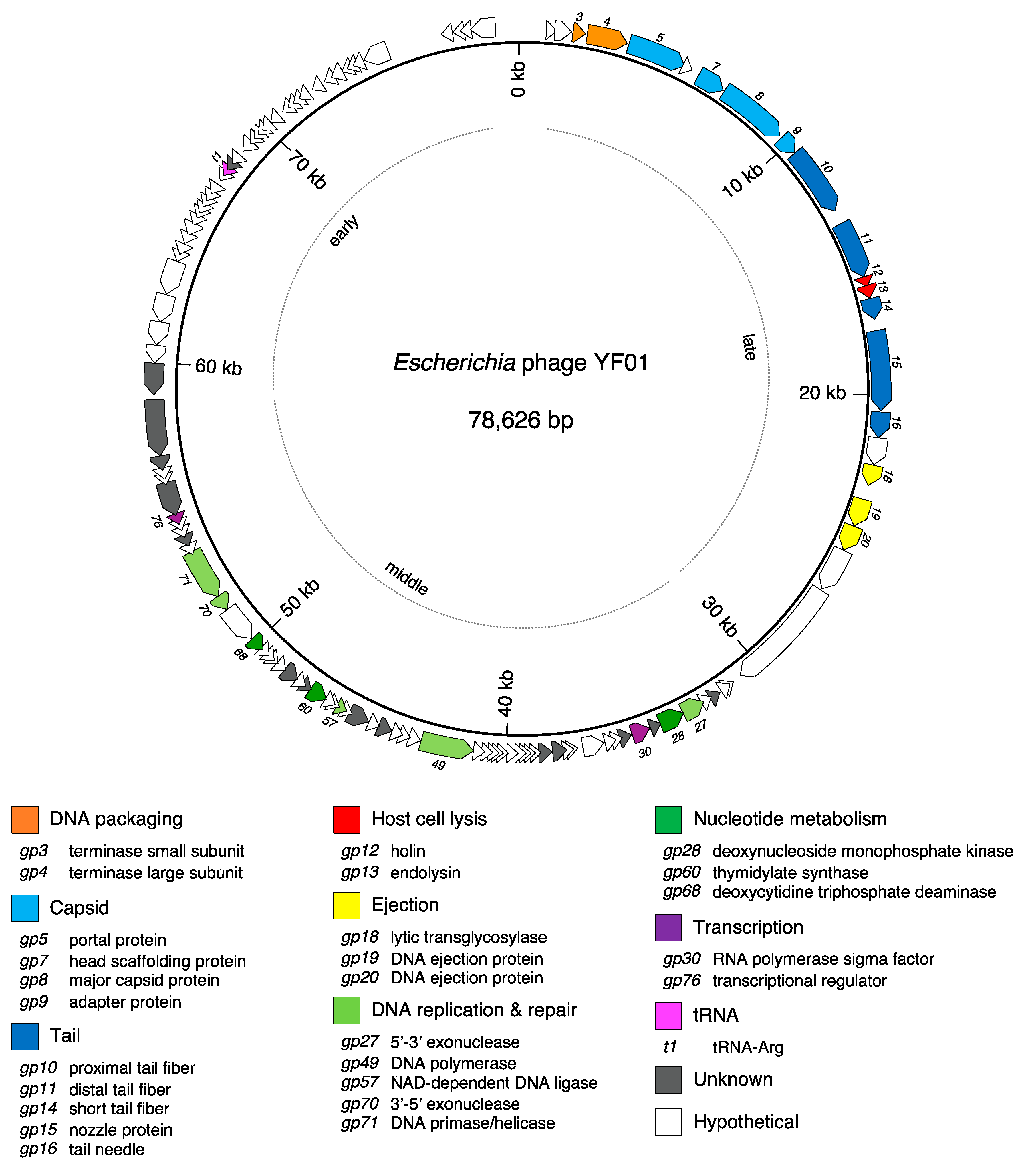
3.3. Genomic Organisation of the YF01 Phage
3.3.1. Genome Termini
3.3.2. Virion Morphogenesis and Lysis
3.3.3. DNA Replication and Nucleotide Metabolism
3.4. The Core Proteome of Kuravirus-like Group Phages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef]
- Michael, I.; Rizzo, L.; McArdell, C.S.; Manaia, C.M.; Merlin, C.; Schwartz, T.; Dagot, C.; Fatta-Kassinos, D. Urban wastewater treatment plants as hotspots for the release of antibiotics in the environment: A review. Water Res. 2013, 47, 957–995. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary rationale for phages as complements of antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Batinovic, S.; Wassef, F.; Knowler, S.A.; Rice, D.T.; Stanton, C.R.; Rose, J.; Tucci, J.; Nittami, T.; Vinh, A.; Drummond, G.R. Bacteriophages in Natural and Artificial Environments. Pathogens 2019, 8, 100. [Google Scholar] [CrossRef]
- Salmond, G.P.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Wang, Q.; Yang, Q.; Wang, R.; Yuan, W.; Yan, L. Responses of bacterial and bacteriophage communities to long-term exposure to antimicrobial agents in wastewater treatment systems. J. Hazard. Mater. 2021, 414, 125486. [Google Scholar] [CrossRef] [PubMed]
- Ewert, D.L.; Paynter, M. Enumeration of bacteriophages and host bacteria in sewage and the activated-sludge treatment process. Appl. Environ. Microbiol. 1980, 39, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Otawa, K.; Lee, S.H.; Yamazoe, A.; Onuki, M.; Satoh, H.; Mino, T. Abundance, diversity, and dynamics of viruses on microorganisms in activated sludge processes. Microb. Ecol. 2007, 53, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, W.-T. Determination of virus abundance, diversity and distribution in a municipal wastewater treatment plant. Water Res. 2009, 43, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Maal, K.B.; Delfan, A.S.; Salmanizadeh, S. Isolation and identification of two novel Escherichia coli bacteriophages and their application in wastewater treatment and coliform’s phage therapy. Jundishapur J. Microbiol. 2015, 8, e14945. [Google Scholar]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 2022, 19, 823–826. [Google Scholar] [CrossRef]
- Luo, J.; Meng, Z.; Xu, X.; Wang, L.; Zhao, K.; Zhu, X.; Qiao, Q.; Ge, Y.; Mao, L.; Cui, L. Systematic benchmarking of nanopore Q20+ kit in SARS-CoV-2 whole genome sequencing. Front. Microbiol. 2022, 13, 973367. [Google Scholar] [CrossRef]
- Vitt, A.R.; Ahern, S.J.; Gambino, M.; Sørensen, M.C.; Brøndsted, L. Genome Sequences of 16 Escherichia coli Bacteriophages Isolated from Wastewater, Pond Water, Cow Manure, and Bird Feces. Microbiol. Resour. Announc. 2022, 11, e00608-22. [Google Scholar] [CrossRef]
- Savalia, D.; Westblade, L.F.; Goel, M.; Florens, L.; Kemp, P.; Akulenko, N.; Pavlova, O.; Padovan, J.C.; Chait, B.T.; Washburn, M.P. Genomic and proteomic analysis of phiEco32, a novel Escherichia coli bacteriophage. J. Mol. Biol. 2008, 377, 774–789. [Google Scholar] [CrossRef]
- Nho, S.-W.; Ha, M.-A.; Kim, K.-S.; Kim, T.-H.; Jang, H.-B.; Cha, I.-S.; Park, S.-B.; Kim, Y.-K.; Jung, T.-S. Complete Genome Sequence of the Bacteriophages ECBP1 and ECBP2 Isolated from Two Different Escherichia coli Strains. J. Virol. 2012, 86, 12439–12440. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Tang, F.; Yao, H.; Lu, C.; Zhang, W. Complete genome sequence of the novel lytic avian pathogenic coliphage NJ01. J. Virol. 2012, 86, 13874–13875. [Google Scholar] [CrossRef]
- Mirzaei, M.K.; Eriksson, H.; Kasuga, K.; Haggård-Ljungquist, E.; Nilsson, A.S. Genomic, proteomic, morphological, and phylogenetic analyses of vB_EcoP_SU10, a podoviridae phage with C3 morphology. PLoS ONE 2014, 9, e116294. [Google Scholar] [CrossRef]
- Stanton, C.R.; Rice, D.T.; Beer, M.; Batinovic, S.; Petrovski, S. Isolation and characterisation of the Bundooravirus genus and phylogenetic investigation of the Salasmaviridae bacteriophages. Viruses 2021, 13, 1557. [Google Scholar] [CrossRef]
- Batinovic, S.; Stanton, C.R.; Rice, D.T.F.; Rowe, B.; Beer, M.; Petrovski, S. Tyroviruses are a new group of temperate phages that infect Bacillus species in soil environments worldwide. BMC Genom. 2022, 23, 777. [Google Scholar] [CrossRef]
- De Coster, W.; D’hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. Infrastructure for a Phage Reference database: Identification of large-scale biases in the current collection of cultured phage genomes. PHAGE 2021, 2, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M. Phylogeny. fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–14. [Google Scholar]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.; Chooi, Y.-H. Clinker & clustermap. js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Rocha, E.P.; Danchin, A. Base composition bias might result from competition for metabolic resources. TRENDS in Genet. 2002, 18, 291–294. [Google Scholar] [CrossRef]
- Shahin, K.; Bao, H.; Zhu, S.; Soleimani-Delfan, A.; He, T.; Mansoorianfar, M.; Wang, R. Bio-control of O157:H7, and colistin-resistant MCR-1-positive Escherichia coli using a new designed broad host range phage cocktail. LWT 2022, 154, 112836. [Google Scholar] [CrossRef]
- Mierlo, J.v.; Hagens, S.; Witte, S.; Klamert, S.; Straat, L.v.d.; Fieseler, L. Complete Genome Sequences of Escherichia coli Phages vB_EcoM-EP75 and vB_EcoP-EP335. Microbiol. Resour. Announc. 2019, 8, e00078-19. [Google Scholar] [CrossRef]
- Manohar, P.; Tamhankar, A.J.; Lundborg, C.S.; Nachimuthu, R. Therapeutic characterization and efficacy of bacteriophage cocktails infecting Escherichia coli, Klebsiella pneumoniae, and Enterobacter species. Front. Microbiol. 2019, 10, 574. [Google Scholar] [CrossRef]
- Manohar, P.; Tamhankar, A.J.; Lundborg, C.S.; Ramesh, N. Isolation, characterization and in vivo efficacy of Escherichia phage myPSH1131. PLoS ONE 2018, 13, e0206278. [Google Scholar] [CrossRef] [PubMed]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [PubMed]
- Korf, I.H.E.; Meier-Kolthoff, J.P.; Adriaenssens, E.M.; Kropinski, A.M.; Nimtz, M.; Rohde, M.; van Raaij, M.J.; Wittmann, J. Still Something to Discover: Novel Insights into Escherichia coli Phage Diversity and Taxonomy. Viruses 2019, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Holt, A.; Saldana, R.; Moreland, R.; Gill, J.J.; Liu, M.; Ramsey, J. Complete Genome Sequence of Escherichia coli Phage Paul. Microbiol. Resour. Announc. 2019, 8, e01093-19. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yan, P.; Xiong, W.; Wang, J.; Liu, X. Genomic characterization of a novel virulent phage infecting Shigella fiexneri and isolated from sewage. Virus Res. 2020, 283, 197983. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.B.; Green, S.I.; Liu, C.G.; Salazar, K.C.; Clark, J.R.; Terwilliger, A.L.; Kaplan, H.B.; Maresso, A.W.; Trautner, B.W.; Ramig, R.F. Constructing and Characterizing Bacteriophage Libraries for Phage Therapy of Human Infections. Front. Microbiol. 2019, 10, 2537. [Google Scholar] [CrossRef] [PubMed]
- Hinkley, T.C.; Garing, S.; Clute-Reinig, N.; Spencer, E.; Jain, P.; Alonzo, L.F.; Ny, A.-L.M.L. Genome Sequences of 38 Bacteriophages Infecting Escherichia coli, Isolated from Raw Sewage. Microbiol. Resour. Announc. 2020, 9, e00909-20. [Google Scholar] [CrossRef]
- Loose, M.; Sáez Moreno, D.; Mutti, M.; Hitzenhammer, E.; Visram, Z.; Dippel, D.; Schertler, S.; Tišáková, L.P.; Wittmann, J.; Corsini, L.; et al. Natural Bred ε2-Phages Have an Improved Host Range and Virulence against Uropathogenic Escherichia coli over Their Ancestor Phages. Antibiotics 2021, 10, 1337. [Google Scholar] [CrossRef]
- Koonjan, S.; Cooper, C.J.; Nilsson, A.S. Complete Genome Sequence of vB_EcoP_SU7, a Podoviridae Coliphage with the Rare C3 Morphotype. Microorganisms 2021, 9, 1576. [Google Scholar] [CrossRef]
- Vera-Mansilla, J.; Sánchez, P.; Silva-Valenzuela, C.A.; Molina-Quiroz, R.C. Isolation and Characterization of Novel Lytic Phages Infecting Multidrug-Resistant Escherichia coli. Microbiol. Spectr. 2022, 10, e01678-21. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Niazi, M.; Florio, T.J.; Yang, R.; Lokareddy, R.K.; Swanson, N.A.; Gillilan, R.E.; Cingolani, G. Biophysical analysis of Pseudomonas-phage PaP3 small terminase suggests a mechanism for sequence-specific DNA-binding by lateral interdigitation. Nucleic Acids Res. 2020, 48, 11721–11736. [Google Scholar] [CrossRef] [PubMed]
- Kala, S.; Cumby, N.; Sadowski, P.D.; Hyder, B.Z.; Kanelis, V.; Davidson, A.R.; Maxwell, K.L. HNH proteins are a widespread component of phage DNA packaging machines. Proc. Natl. Acad. Sci. USA 2014, 111, 6022–6027. [Google Scholar] [CrossRef]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell 2004, 16, 11–21. [Google Scholar] [CrossRef]
- Condreay, J.P.; Wright, S.E.; Molineux, I.J. Nucleotide sequence and complementation studies of the gene 10 region of bacteriophage T3. J. Mol. Biol. 1989, 207, 555–561. [Google Scholar] [CrossRef]
- Dunn, J.J.; Studier, F.W. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 1983, 166, 477–535. [Google Scholar] [CrossRef]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef]
- Šiborová, M.; Füzik, T.; Procházková, M.; Nováček, J.; Benešík, M.; Nilsson, A.S.; Plevka, P. Tail proteins of phage SU10 reorganize into the nozzle for genome delivery. Nat. Commun. 2022, 13, 5622. [Google Scholar] [CrossRef]
- Pavlova, O.; Lavysh, D.; Klimuk, E.; Djordjevic, M.; Ravcheev, D.A.; Gelfand, M.S.; Severinov, K.; Akulenko, N. Temporal regulation of gene expression of the Escherichia coli bacteriophage phiEco32. J. Mol. Biol. 2012, 416, 389–399. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.T.; Ku, H.; Low, Y.P.; Batinovic, S.; Kabwe, M.; Petrovski, S.; Tucci, J. Characterization of novel lytic bacteriophages of Achromobacter marplantensis isolated from a pneumonia patient. Viruses 2020, 12, 1138. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.; Kabwe, M.; Chan, H.T.; Stanton, C.; Petrovski, S.; Batinovic, S.; Tucci, J. Novel Drexlerviridae bacteriophage KMI8 with specific lytic activity against Klebsiella michiganensis and its biofilms. PLoS ONE 2021, 16, e0257102. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Host | Size (nt) | GC % | CDS 1 | tRNA 2 | Geography | Source | Year | Accession | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| YF01 | E. coli | 78,626 | 42.1 | 121 | 1 | Japan | Wastewater | 2021 | OQ025076 | This study |
| phiEco32 | E. coli | 77,554 | 42.3 | 124 | 1 | USA | Water | 2004 | EU330206 | [17] |
| ECBP2 | E. coli | 77,315 | 42.4 | 119 | 1 | South Korea | Unknown | 2012 3 | JX415536 | [18] |
| NJ01 | E. coli | 77,448 | 42 | 132 | 1 | China | Animal | 2012 3 | JX867715 | [19] |
| KBNP1711 | E. coli | 76,184 | 42.4 | 124 | 1 | South Korea | Unknown | 2013 3 | KF981730 | |
| SU10 | E. coli | 77,327 | 42.1 | 124 | 1 | Sweden | Unknown | 2014 3 | KM044272 | [20] |
| 172-1 | E. coli | 77,266 | 42 | 128 | 1 | China | Animal feces | 2014 3 | KP308307 | |
| EK010 | E. coli | 78,078 | 42.1 | 120 | 1 | China | Wastewater | 2020 3 | LC553734 | [50] |
| O18-011 | E. coli | 75,646 | 42.1 | 122 | 1 | China | Unknown | 2020 3 | LC553735 | [50] |
| LAMP | E. coli | 68,521 | 42.2 | 96 | 1 | Russia | Animal feces | 2017 3 | MG673519 | |
| EP335 | E. coli | 76,622 | 42.5 | 123 | 1 | Netherlands | Wastewater | 2018 3 | MG748548 | [51] |
| myPSH2311 | E. coli | 68,712 | 42.3 | 118 | 0 | India | Wastewater | 2018 3 | MG976803 | [52] |
| myPSH1131 | E. coli | 76,163 | 42.4 | 129 | 0 | India | Water | 2018 3 | MG983840 | [53] |
| NC-B | E. coli | 76,641 | 42.1 | 116 | 1 | USA | Human feces | 2019 3 | MK310183 | [54] |
| WFI101126 | E. coli | 77,307 | 42.1 | 130 | 1 | Germany | Sewage | 2015 | MK373770 | [55] |
| Paul | E. coli | 79,429 | 42 | 124 | 1 | USA | Water | 2018 | MN045231 | [56] |
| SGF2 | S. flexneri | 76,964 | 42.3 | 118 | 1 | China | Water | 2019 3 | MN148435 | [57] |
| ES17 | E. coli | 75,007 | 42.1 | 120 | 1 | USA | Sewage | 2018 | MN508615 | [58] |
| EcoN5 | E. coli | 76,083 | 42.1 | 128 | 1 | Colombia | Unknown | 2019 3 | MN715356 | |
| PGN6866 | E. coli | 78,549 | 42.3 | 129 | 1 | India | Sewage | 2020 3 | MT127620 | |
| MN03 | E. coli | 77,187 | 42.2 | 124 | 1 | Bangladesh | Water | 2017 | MT129653 | |
| MN05 | E. coli | 76,899 | 42.2 | 126 | 1 | Bangladesh | Water | 2017 | MT129655 | |
| TH06 | E. coli | 77,678 | 42.1 | 123 | 1 | USA | Wastewater | 2020 3 | MT446386 | [59] |
| TH34 | E. coli | 77,944 | 42.3 | 120 | 1 | USA | Wastewater | 2020 3 | MT446407 | [59] |
| TH38 | E. coli | 81,552 | 42.2 | 132 | 1 | USA | Wastewater | 2020 3 | MT446410 | [59] |
| TH42 | E. coli | 77,284 | 42.3 | 118 | 0 | USA | Wastewater | 2020 3 | MT446413 | [59] |
| TH43 | E. coli | 77,980 | 42.4 | 118 | 1 | USA | Wastewater | 2020 3 | MT446414 | [59] |
| DE5 | E. coli | 77,305 | 42.1 | 125 | 1 | China | Unknown | 2021 3 | MW741821 | |
| 101114BS3 | E. coli | 75,747 | 42 | 113 | 1 | Austria | Wastewater | 2018 | MZ234015 | [60] |
| 101114UKE3 | E. coli | 75,747 | 42 | 113 | 1 | Austria | Wastewater | 2018 | MZ234017 | [60] |
| CHD5UKE1 | E. coli | 77,359 | 42.2 | 123 | 1 | Austria | Wastewater | 2018 | MZ234028 | [60] |
| SU7 | E. coli | 76,626 | 42.1 | 117 | 1 | Sweden | Wastewater | 2016 | MZ342906 | [61] |
| IME267 | E. coli | 76,631 | 42 | 117 | 1 | China | Unknown | 2021 3 | MZ398243 | |
| IMEP8 | E. coli | 75,809 | 42.1 | 118 | 1 | China | Animal milk | 2021 | MZ648214 | |
| MLP3 | E. coli | 76,234 | 42.1 | 115 | 1 | Chile | Water | 2019 | OK148440 | [62] |
| E20-1 | E. coli | 77,938 | 42.2 | 122 | 1 | China | Wastewater | 2018 | OP293233 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batinovic, S.; Fujii, Y.; Nittami, T. Expansion of Kuravirus-like Phage Sequences within the Past Decade, including Escherichia Phage YF01 from Japan, Prompt the Creation of Three New Genera. Viruses 2023, 15, 506. https://doi.org/10.3390/v15020506
Batinovic S, Fujii Y, Nittami T. Expansion of Kuravirus-like Phage Sequences within the Past Decade, including Escherichia Phage YF01 from Japan, Prompt the Creation of Three New Genera. Viruses. 2023; 15(2):506. https://doi.org/10.3390/v15020506
Chicago/Turabian StyleBatinovic, Steven, Yugo Fujii, and Tadashi Nittami. 2023. "Expansion of Kuravirus-like Phage Sequences within the Past Decade, including Escherichia Phage YF01 from Japan, Prompt the Creation of Three New Genera" Viruses 15, no. 2: 506. https://doi.org/10.3390/v15020506
APA StyleBatinovic, S., Fujii, Y., & Nittami, T. (2023). Expansion of Kuravirus-like Phage Sequences within the Past Decade, including Escherichia Phage YF01 from Japan, Prompt the Creation of Three New Genera. Viruses, 15(2), 506. https://doi.org/10.3390/v15020506