

Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging

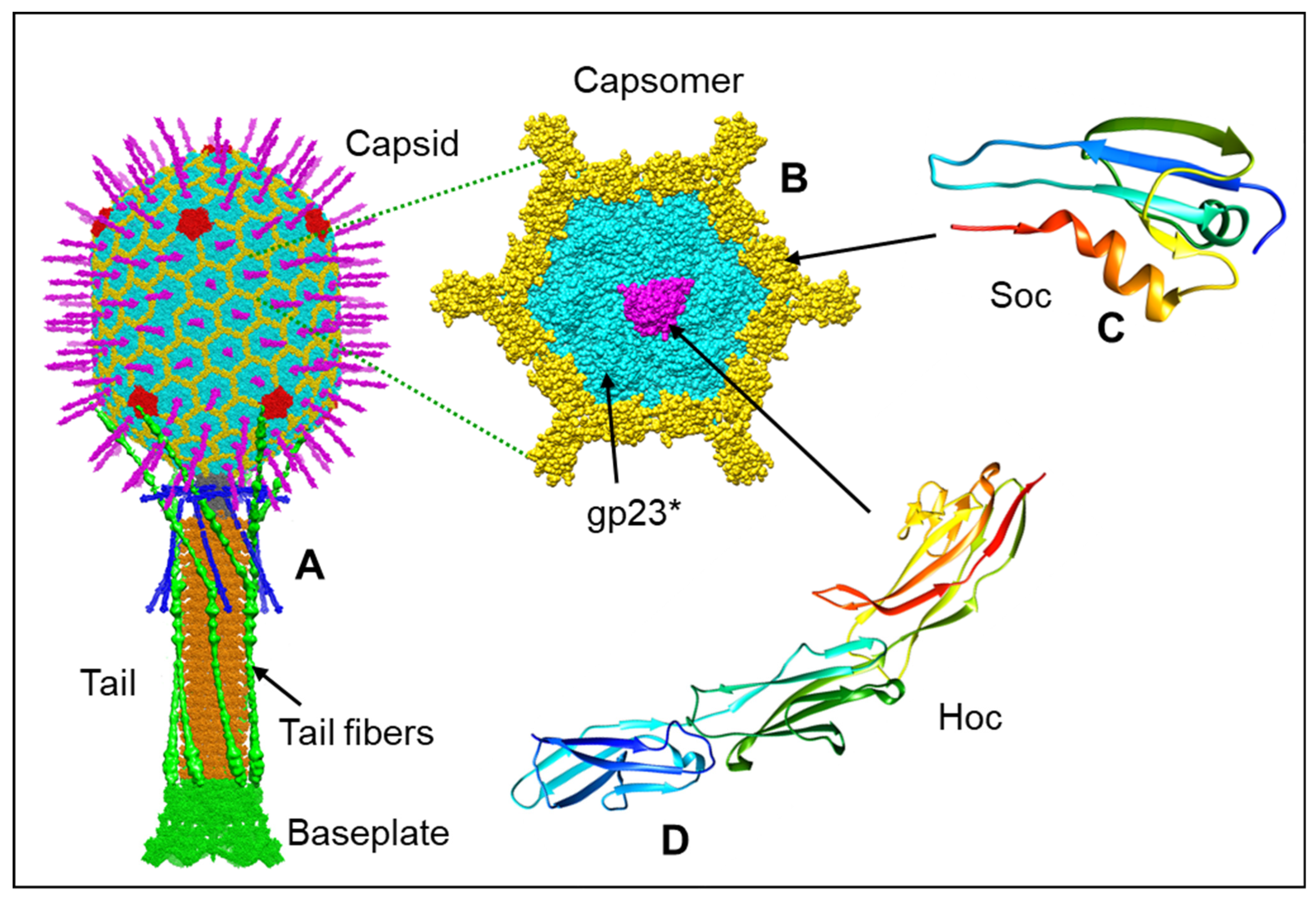{kind=link}
{kind=link}
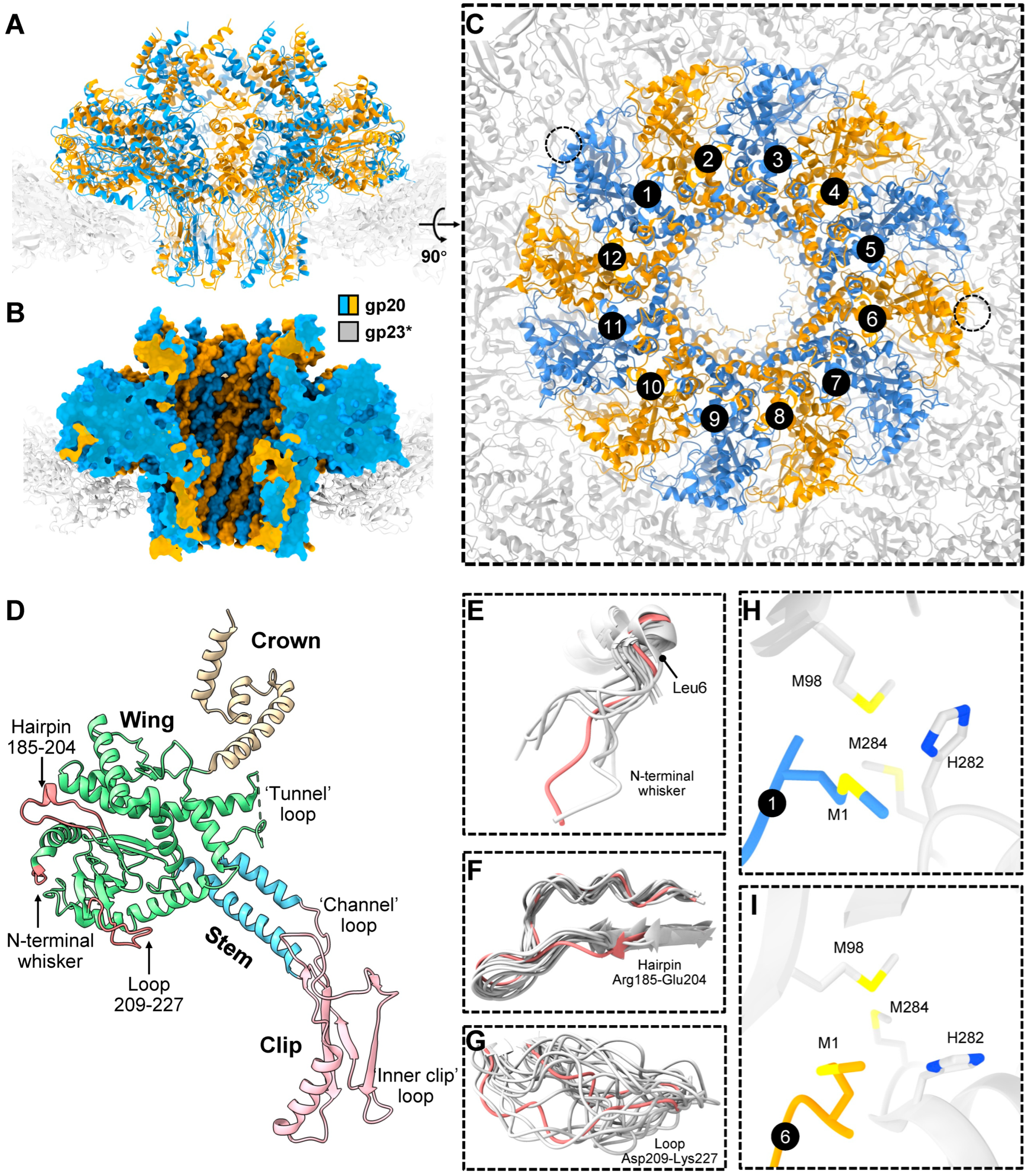{kind=link}
{kind=link}
{kind=link}
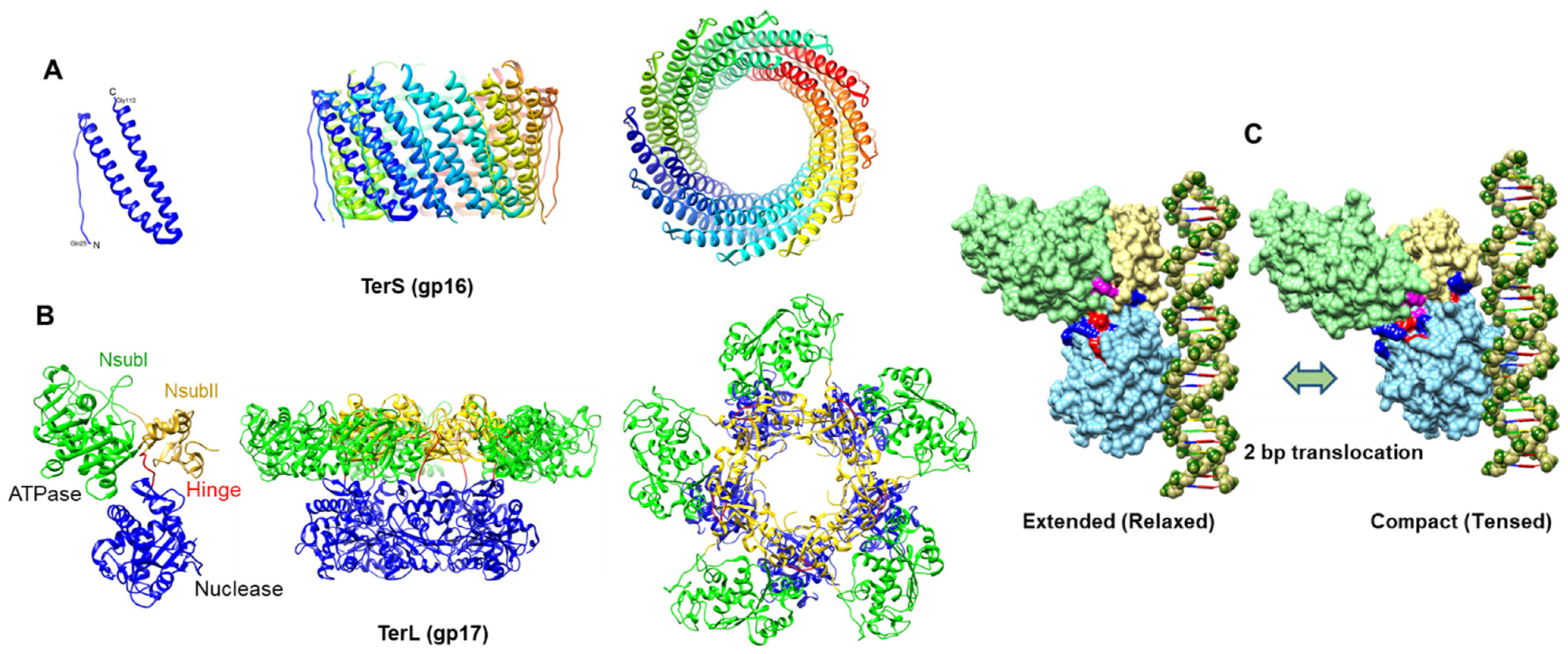{kind=link}
{kind=link}
Abstract
:1. Introduction
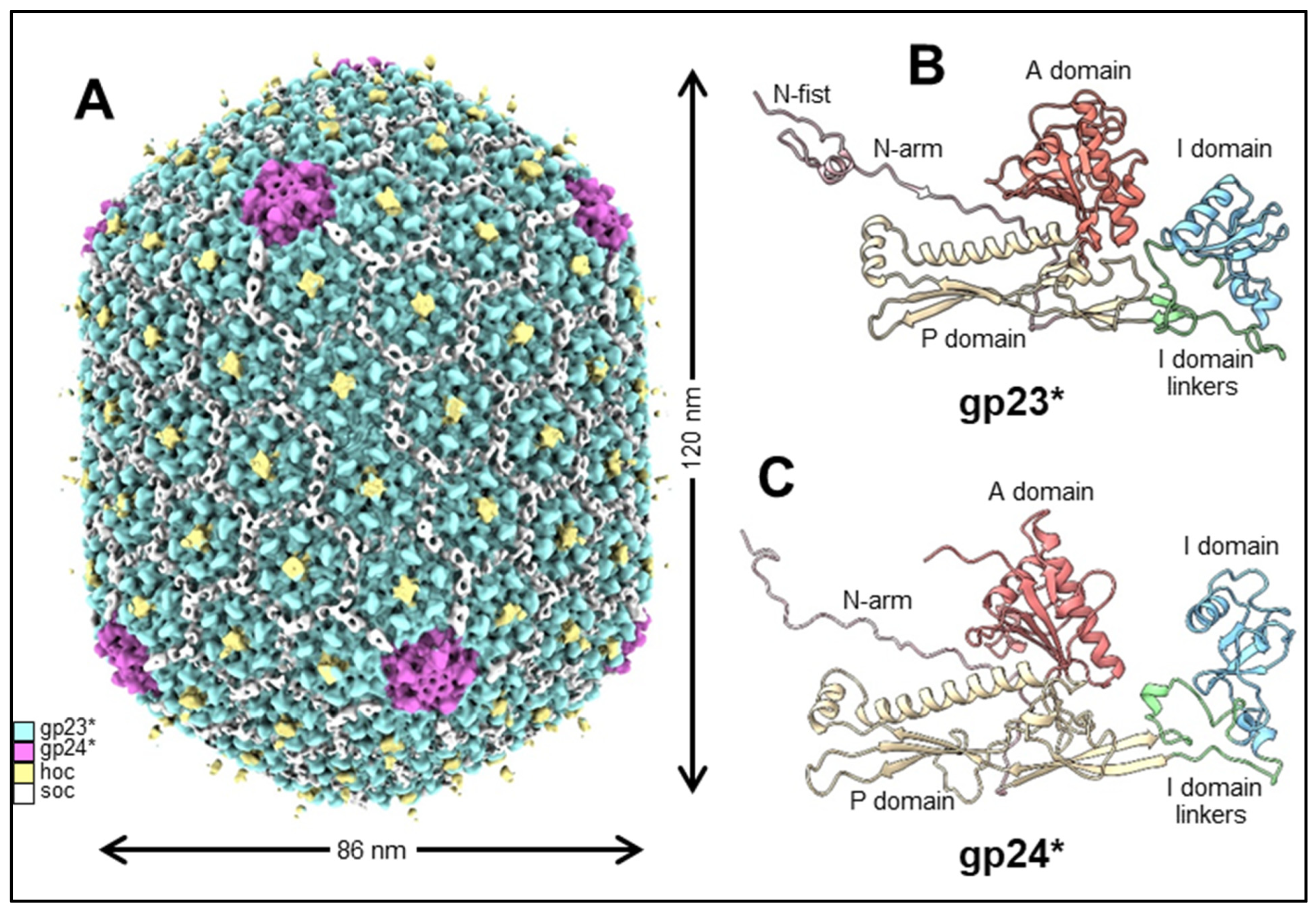
2. Architecture of T4 Head
2.1. Capsid Shell
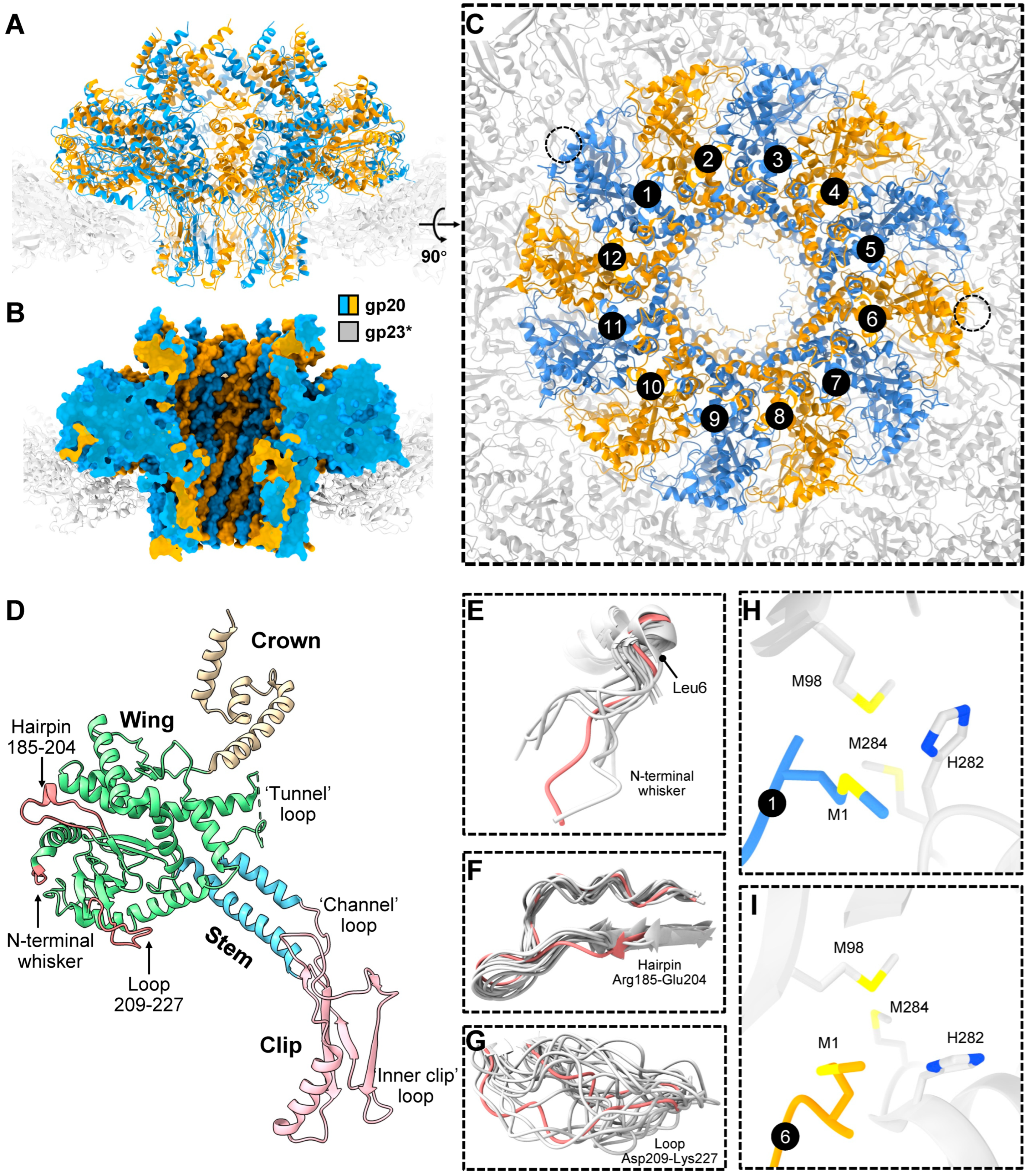
2.2. Portal
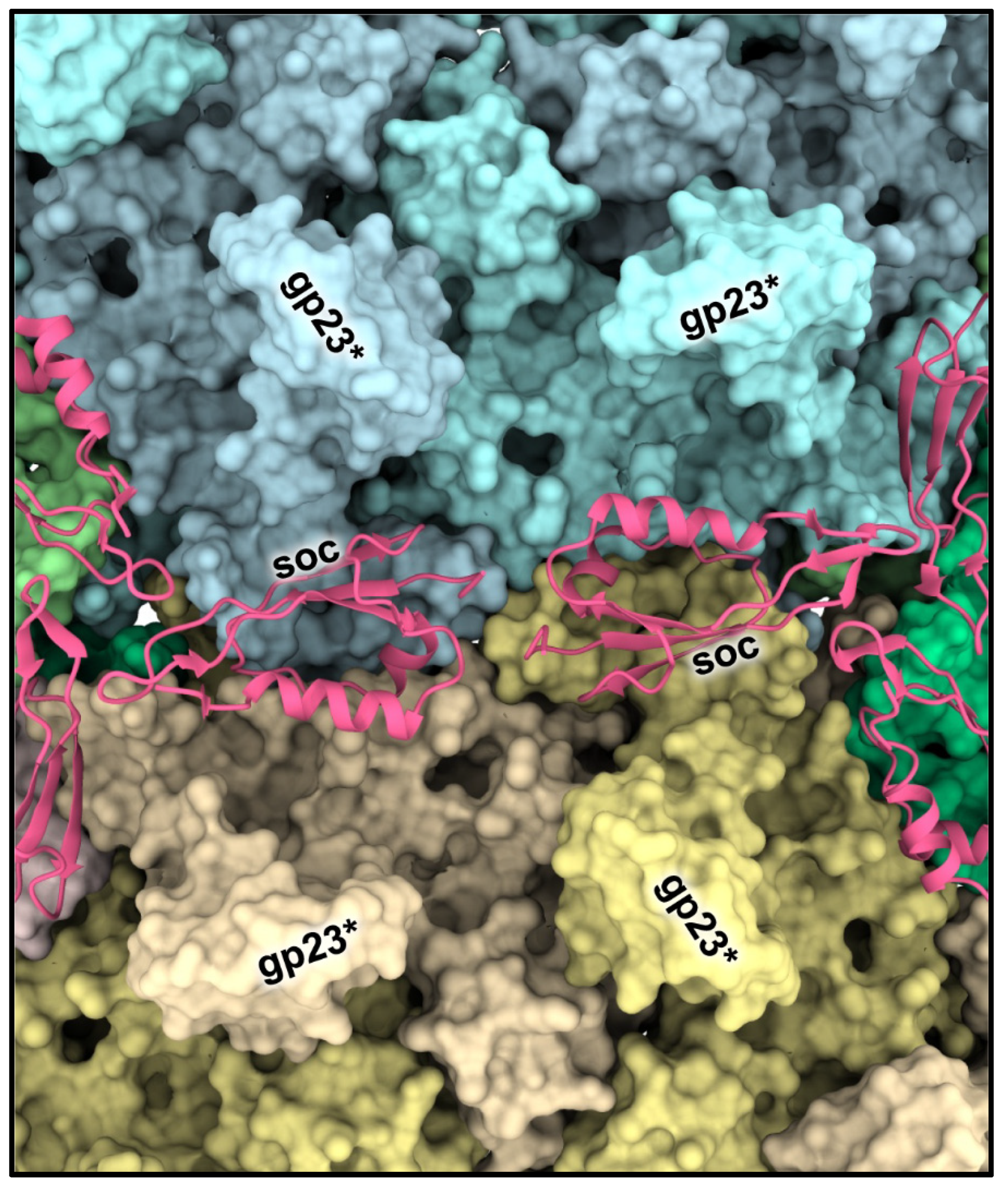
2.3. Decoration Proteins
3. Head Assembly
3.1. Assembly Pathway and Maturation
3.2. Expansion
3.3. Length Control
4. Genome Packaging
4.1. TerS
4.2. TerL
4.2.1. ATPase
4.2.2. Nuclease
4.2.3. Translocase
4.3. Packaging Motor
4.4. Packaging Dynamics and Mechanism
4.4.1. Electrostatic Force Generation
4.4.2. DNA Structural Transitions
4.4.3. Continuous Burst
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rao, V.B.; Black, L.W. DNA packaging of bacteriophage T4 proheads in vitro. Evidence that prohead expansion is not coupled to DNA packaging. J. Mol. Biol. 1985, 185, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Black, L.W. Cloning, overexpression and purification of the terminase proteins gp16 and gp17 of bacteriophage T4. Construction of a defined in-vitro DNA packaging system using purified terminase proteins. J. Mol. Biol. 1988, 200, 475–488. [Google Scholar]
- Rao, V.B.; Black, L.W. Structure and assembly of bacteriophage T4 head. Virol. J. 2010, 7, 356. [Google Scholar] [CrossRef]
- Black, L.W.; Rao, V.B. Structure, assembly, and DNA packaging of the bacteriophage T4 head. Adv. Virus Res. 2012, 82, 119–153. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Black, L.W. DNA packaging in bacteriophage T4. In Viral Genome Packaging Machines: Genetics, Structure and Mechanism; Catalano, C.E., Ed.; Landes Bioscience: Georgetown, TX, USA, 2005; pp. 40–58. [Google Scholar]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef]
- Hendrix, R.W. Bacteriophage genomics. Curr. Opin. Microbiol. 2003, 6, 506–511. [Google Scholar] [CrossRef]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef]
- Tao, P.; Zhu, J.; Mahalingam, M.; Batra, H.; Rao, V.B. Bacteriophage T4 nanoparticles for vaccine delivery against infectious diseases. Adv. Drug. Deliv. Rev. 2019, 145, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Zhu, J. Bacteriophage T4 as a nanovehicle for delivery of genes and therapeutics into human cells. Curr. Opin. Virol. 2022, 55, 101255. [Google Scholar] [CrossRef]
- Zhu, J.; Ananthaswamy, N.; Jain, S.; Batra, H.; Tang, W.C.; Rao, V.B. CRISPR Engineering of Bacteriophage T4 to Design Vaccines Against SARS-CoV-2 and Emerging Pathogens. Methods. Mol. Biol. 2022, 2410, 209–228. [Google Scholar] [CrossRef]
- Eiserling, F.A.; Black, L.W. Pathways in T4 morphogenesis. In Molecular Biology of Bacteriophage T4; Karam, J.D., Drake, J.W., Kreuzer, K.N., Mosig, G., Hall, D.H., Eiserling, F.A., Black, L.W., Spicer, E.K., Kutter, E., Carlson, C., et al., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 209–212. [Google Scholar]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef] [Green Version]
- Fokine, A.; Chipman, P.R.; Leiman, P.G.; Mesyanzhinov, V.V.; Rao, V.B.; Rossmann, M.G. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. USA 2004, 101, 6003–6008. [Google Scholar] [CrossRef]
- Fang, Q.; Tang, W.C.; Fokine, A.; Mahalingam, M.; Shao, Q.; Rossmann, M.G.; Rao, V.B. Structures of a large prolate virus capsid in unexpanded and expanded states generate insights into the icosahedral virus assembly. Proc. Natl. Acad. Sci. USA 2022, 119, e2203272119. [Google Scholar] [CrossRef]
- Black, L.W.; Showe, M.K.; Steven, A.C. Morphogenesis of the T4 head. In Molecular Biology of Bacteriophage T4; Karam, J.D., Ed.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 218–258. [Google Scholar]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. 2003, 60, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Sun, L.; Zhang, Z.; Fokine, A.; Padilla-Sanchez, V.; Hanein, D.; Jiang, W.; Rossmann, M.G.; Rao, V.B. Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. Proc. Natl. Acad. Sci. USA 2017, 114, E8184–E8193. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Battisti, A.J.; Kostyuchenko, V.A.; Black, L.W.; Rossmann, M.G. Cryo-EM structure of a bacteriophage T4 gp24 bypass mutant: The evolution of pentameric vertex proteins in icosahedral viruses. J. Struct. Biol. 2006, 154, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Choi, K.H.; Koti, J.S.; Chipman, P.R.; Anderson, D.L.; Rossmann, M.G. Conservation of the capsid structure in tailed dsDNA bacteriophages: The pseudoatomic structure of phi29. Mol. Cell 2005, 18, 149–159. [Google Scholar] [CrossRef]
- Parent, K.N.; Khayat, R.; Tu, L.H.; Suhanovsky, M.M.; Cortines, J.R.; Teschke, C.M.; Johnson, J.E.; Baker, T.S. P22 coat protein structures reveal a novel mechanism for capsid maturation: Stability without auxiliary proteins or chemical crosslinks. Structure 2010, 18, 390–401. [Google Scholar] [CrossRef]
- Chen, D.H.; Baker, M.L.; Hryc, C.F.; DiMaio, F.; Jakana, J.; Wu, W.; Dougherty, M.; Haase-Pettingell, C.; Schmid, M.F.; Jiang, W.; et al. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. USA 2011, 108, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Duda, R.L.; Teschke, C.M. The amazing HK97 fold: Versatile results of modest differences. Curr. Opin. Virol. 2019, 36, 9–16. [Google Scholar] [CrossRef]
- Black, L.W.; Newcomb, W.W.; Boring, J.W.; Brown, J.C. Ion etching bacteriophage T4: Support for a spiral-fold model of packaged DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 7960–7964. [Google Scholar] [CrossRef]
- Black, L.W.; Thomas, J.A. Condensed genome structure. Adv. Exp. Med. Biol. 2012, 726, 469–487. [Google Scholar] [CrossRef]
- Johnson, K.; Condie, B.; Mooney, D.T.; Doermann, A.H. Mutations that eliminate the requirement for the vertex protein in bacteriophage T4 capsid assembly. J. Mol. Biol. 1992, 224, 601–611. [Google Scholar] [CrossRef]
- Driedonks, R.A.; Engel, A.; tenHeggeler, B.; van Driel. Gene 20 product of bacteriophage T4 its purification and structure. J. Mol. Biol. 1981, 152, 641–662. [Google Scholar] [CrossRef]
- Rao, V.B.; Feiss, M. Mechanisms of DNA Packaging by Large Double-Stranded DNA Viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Kondabagil, K.; Draper, B.; Alam, T.I.; Bowman, V.D.; Zhang, Z.; Hegde, S.; Fokine, A.; Rossmann, M.G.; Rao, V.B. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 2008, 135, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, X.; Gao, S.; Rao, P.A.; Padilla-Sanchez, V.; Chen, Z.; Sun, S.; Xiang, Y.; Subramaniam, S.; Rao, V.B.; et al. Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat. Commun. 2015, 6, 7548. [Google Scholar] [CrossRef]
- Fang, Q.; Tang, W.C.; Tao, P.; Mahalingam, M.; Fokine, A.; Rossmann, M.G.; Rao, V.B. Structural morphing in a symmetry-mismatched viral vertex. Nat. Commun. 2020, 11, 1713. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Fokine, A.; Fang, Q. The remarkable viral portal vertex: Structure and a plausible model for mechanism. Curr. Opin. Virol. 2021, 51, 65–73. [Google Scholar] [CrossRef]
- Dedeo, C.L.; Cingolani, G.; Teschke, C.M. Portal Protein: The Orchestrator of Capsid Assembly for the dsDNA Tailed Bacteriophages and Herpesviruses. Annu. Rev. Virol. 2019, 6, 141–160. [Google Scholar] [CrossRef]
- Liu, Y.T.; Jih, J.; Dai, X.; Bi, G.Q.; Zhou, Z.H. Cryo-EM structures of herpes simplex virus type 1 portal vertex and packaged genome. Nature 2019, 570, 257–261. [Google Scholar] [CrossRef]
- Fokine, A.; Zhang, Z.; Kanamaru, S.; Bowman, V.D.; Aksyuk, A.A.; Arisaka, F.; Rao, V.B.; Rossmann, M.G. The molecular architecture of the bacteriophage T4 neck. J. Mol. Biol. 2013, 425, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Fokine, A.; O’Donnell, E.; Rao, V.B.; Rossmann, M.G. Structure of the small outer capsid protein, Soc: A clamp for stabilizing capsids of T4-like phages. J. Mol. Biol. 2010, 395, 728–741. [Google Scholar] [CrossRef]
- Fokine, A.; Islam, M.Z.; Zhang, Z.; Bowman, V.D.; Rao, V.B.; Rossmann, M.G. Structure of the three N-terminal immunoglobulin domains of the highly immunogenic outer capsid protein from a T4-like bacteriophage. J. Virol. 2011, 85, 8141–8148. [Google Scholar] [CrossRef]
- Ishii, T.; Yanagida, M. The two dispensable structural proteins (soc and hoc) of the T4 phage capsid; their purification and properties, isolation and characterization of the defective mutants, and their binding with the defective heads in vitro. J. Mol. Biol. 1977, 109, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Sathaliyawala, T.; Islam, M.Z.; Li, Q.; Fokine, A.; Rossmann, M.G.; Rao, V.B. Functional analysis of the highly antigenic outer capsid protein, Hoc, a virus decoration protein from T4-like bacteriophages. Mol. Microbiol. 2010, 77, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, K.; Switała-Jeleń, K.; Opolski, A.; Górski, A. Possible association between phages, Hoc protein, and the immune system. Arch. Virol. 2006, 151, 209–215. [Google Scholar] [CrossRef]
- Dabrowska, K.; Zembala, M.; Boratynski, J.; Switala-Jelen, K.; Wietrzyk, J.; Opolski, A.; Szczaurska, K.; Kujawa, M.; Godlewska, J.; Gorski, A. Hoc protein regulates the biological effects of T4 phage in mammals. Arch. Microbiol. 2007, 187, 489–498. [Google Scholar] [CrossRef]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, W.H.; Kett, C.; Cooper, O.; Müseler, D.; Zhang, Y.; Bamert, R.S.; Patwa, R.; Woods, L.C.; Devendran, C.; Korneev, D.; et al. Bacteriophages evolve enhanced persistence to a mucosal surface. Proc. Natl. Acad. Sci. USA 2022, 119, e2116197119. [Google Scholar] [CrossRef] [PubMed]
- Lander, G.C.; Evilevitch, A.; Jeembaeva, M.; Potter, C.S.; Carragher, B.; Johnson, J.E. Bacteriophage lambda stabilization by auxiliary protein gpD: Timing, location, and mechanism of attachment determined by cryo-EM. Structure 2008, 16, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hardies, S.C.; Fokine, A.; Klose, T.; Jiang, W.; Cho, B.C.; Rossmann, M.G. Structure of the Marine Siphovirus TW1: Evolution of Capsid-Stabilizing Proteins and Tail Spikes. Structure 2018, 26, 238–248.e233. [Google Scholar] [CrossRef]
- Ren, Z.J.; Lewis, G.K.; Wingfield, P.T.; Locke, E.G.; Steven, A.C.; Black, L.W. Phage display of intact domains at high copy number: A system based on SOC, the small outer capsid protein of bacteriophage T4. Protein Sci. 1996, 5, 1833–1843. [Google Scholar] [CrossRef]
- Jiang, J.; Abu-Shilbayeh, L.; Rao, V.B. Display of a PorA peptide from Neisseria meningitidis on the bacteriophage T4 capsid surface. Infect. Immun. 1997, 65, 4770–4777. [Google Scholar] [CrossRef]
- Li, M.; Guo, P.; Chen, C.; Feng, H.; Zhang, W.; Gu, C.; Wen, G.; Rao, V.B.; Tao, P. Bacteriophage T4 Vaccine Platform for Next-Generation Influenza Vaccine Development. Front. Immunol. 2021, 12, 745625. [Google Scholar] [CrossRef]
- Fokine, A.; Bowman, V.D.; Battisti, A.J.; Li, Q.; Chipman, P.R.; Rao, V.B.; Rossmann, M.G. Cryo-electron microscopy study of bacteriophage T4 displaying anthrax toxin proteins. Virology 2007, 367, 422–427. [Google Scholar] [CrossRef]
- Zhu, J.; Jain, S.; Sha, J.; Batra, H.; Ananthaswamy, N.; Kilgore, P.B.; Hendrix, E.K.; Hosakote, Y.M.; Wu, X.; Olano, J.P.; et al. A Bacteriophage-Based, Highly Efficacious, Needle- and Adjuvant-Free, Mucosal COVID-19 Vaccine. mBio 2022, 13, e0182222. [Google Scholar] [CrossRef]
- Kuhn, A.; Thomas, J.A. The Beauty of Bacteriophage T4 Research: Lindsay W. Black and the T4 Head Assembly. Viruses 2022, 14, 700. [Google Scholar] [CrossRef]
- Quinten, T.A.; Kuhn, A. Membrane interaction of the portal protein gp20 of bacteriophage T4. J. Virol. 2012, 86, 11107–11114. [Google Scholar] [CrossRef] [Green Version]
- Bair, C.L.; Rifat, D.; Black, L.W. Exclusion of glucosyl-hydroxymethylcytosine DNA containing bacteriophages is overcome by the injected protein inhibitor IPI*. J. Mol. Biol. 2007, 366, 779–789. [Google Scholar] [CrossRef]
- Rifat, D.; Wright, N.T.; Varney, K.M.; Weber, D.J.; Black, L.W. Restriction endonuclease inhibitor IPI* of bacteriophage T4: A novel structure for a dedicated target. J. Mol. Biol. 2008, 375, 720–734. [Google Scholar] [CrossRef]
- Koch, T.; Raudonikiene, A.; Wilkens, K.; Rüger, W. Overexpression, purification, and characterization of the ADP-ribosyltransferase (gpAlt) of bacteriophage T4: ADP-ribosylation of E. coli RNA polymerase modulates T4 “early” transcription. Gene. Expr. 1995, 4, 253–264. [Google Scholar]
- Mullaney, J.M.; Black, L.W. Capsid targeting sequence targets foreign proteins into bacteriophage T4 and permits proteolytic processing. J. Mol. Biol. 1996, 261, 372–385. [Google Scholar] [CrossRef]
- Mullaney, J.M.; Black, L.W. Bacteriophage T4 capsid packaging and unpackaging of DNA and proteins. Methods Mol. Biol. 2014, 1108, 69–85. [Google Scholar] [CrossRef]
- Fokine, A.; Rossmann, M.G. Common Evolutionary Origin of Procapsid Proteases, Phage Tail Tubes, and Tubes of Bacterial Type VI Secretion Systems. Structure 2016, 24, 1928–1935. [Google Scholar] [CrossRef]
- McNicol, L.A.; Simon, L.E. A mutation which bypasses the requirement for p24 in bacteriophage T4 capsid morphogenesis. J. Mol. Biol. 1977, 116, 261–283. [Google Scholar] [CrossRef]
- Ghosh-Kumar, M.; Alam, T.I.; Draper, B.; Stack, J.D.; Rao, V.B. Regulation by interdomain communication of a headful packaging nuclease from bacteriophage T4. Nucleic Acids Res. 2011, 39, 2742–2755. [Google Scholar] [CrossRef]
- Ray, K.; Oram, M.; Ma, J.; Black, L.W. Portal control of viral prohead expansion and DNA packaging. Virology 2009, 391, 44–50. [Google Scholar] [CrossRef]
- Steven, A.C.; Carrascosa, J.L. Proteolytic cleavage and structural transformation: Their relationship in bacteriophage T4 capsid maturation. J. Supramol. Struct. 1979, 10, 1–11. [Google Scholar] [CrossRef]
- Müller, M.; Mesyanzhinov, V.V.; Aebi, U. In vitro maturation of prehead-like bacteriophage T4 polyheads: Structural changes accompanying proteolytic cleavage and lattice expansion. J. Struct. Biol. 1994, 112, 199–215. [Google Scholar] [CrossRef]
- Ray, K.; Ma, J.; Oram, M.; Lakowicz, J.R.; Black, L.W. Single-molecule and FRET fluorescence correlation spectroscopy analyses of phage DNA packaging: Colocalization of packaged phage T4 DNA ends within the capsid. J. Mol. Biol. 2010, 395, 1102–1113. [Google Scholar] [CrossRef]
- Mo, Y.; Keller, N.; delToro, D.; Ananthaswamy, N.; Harvey, S.C.; Rao, V.B.; Smith, D.E. Function of a viral genome packaging motor from bacteriophage T4 is insensitive to DNA sequence. Nucleic Acids Res. 2020, 48, 11602–11614. [Google Scholar] [CrossRef]
- Lane, T.; Eiserling, F. Genetic control of capsid length in bacteriophage T4. VII. A model of length regulation based on DNA size. J. Struct. Biol. 1990, 104, 9–23. [Google Scholar] [CrossRef]
- Doermann, A.H.; Pao, A.; Jackson, P. Genetic control of capsid length in bacteriophage T4: Clustering of ptg mutations in gene 23. J. Virol. 1987, 61, 2823–2827. [Google Scholar] [CrossRef]
- Mooney, D.T.; Stockard, J.; Parker, M.L.; Doermann, A.H. Genetic control of capsid length in bacteriophage T4: DNA sequence analysis of petite and petite/giant mutants. J. Virol. 1987, 61, 2828–2834. [Google Scholar] [CrossRef]
- Black, L.W. Old, new, and widely true: The bacteriophage T4 DNA packaging mechanism. Virology 2015, 479–480, 650–656. [Google Scholar] [CrossRef]
- Casjens, S.R. The DNA-packaging nanomotor of tailed bacteriophages. Nat. Rev. Microbiol. 2011, 9, 647–657. [Google Scholar] [CrossRef]
- Chemla, Y.R.; Smith, D.E. Single-molecule studies of viral DNA packaging. Adv. Exp. Med. Biol. 2012, 726, 549–584. [Google Scholar] [CrossRef]
- Catalano, C.E.; Morais, M.C. Viral genome packaging machines: Structure and enzymology. Enzymes 2021, 50, 369–413. [Google Scholar] [CrossRef]
- Oliveira, L.; Tavares, P.; Alonso, J.C. Headful DNA packaging: Bacteriophage SPP1 as a model system. Virus Res. 2013, 173, 247–259. [Google Scholar] [CrossRef]
- Black, L.W. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 1989, 43, 267–292. [Google Scholar] [CrossRef]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef]
- Leffers, G.; Rao, V.B. Biochemical characterization of an ATPase activity associated with the large packaging subunit gp17 from bacteriophage T4. J. Biol. Chem. 2000, 275, 37127–37136. [Google Scholar] [CrossRef]
- Lin, H.; Simon, M.N.; Black, L.W. Purification and characterization of the small subunit of phage T4 terminase, gp16, required for DNA packaging. J. Biol. Chem. 1997, 272, 3495–3501. [Google Scholar] [CrossRef]
- Lin, H.; Black, L.W. DNA requirements in vivo for phage T4 packaging. Virology 1998, 242, 118–127. [Google Scholar] [CrossRef]
- Al-Zahrani, A.S.; Kondabagil, K.; Gao, S.; Kelly, N.; Ghosh-Kumar, M.; Rao, V.B. The small terminase, gp16, of bacteriophage T4 is a regulator of the DNA packaging motor. J. Biol. Chem. 2009, 284, 24490–24500. [Google Scholar] [CrossRef]
- Gao, S.; Rao, V.B. Specificity of interactions among the DNA-packaging machine components of T4-related bacteriophages. J. Biol. Chem. 2011, 286, 3944–3956. [Google Scholar] [CrossRef]
- Mitchell, M.S.; Matsuzaki, S.; Imai, S.; Rao, V.B. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 2002, 30, 4009–4021. [Google Scholar] [CrossRef]
- van Duijn, E. Current limitations in native mass spectrometry based structural biology. J. Am. Soc. Mass Spectrom. 2010, 21, 971–978. [Google Scholar] [CrossRef]
- Kondabagil, K.R.; Rao, V.B. A critical coiled coil motif in the small terminase, gp16, from bacteriophage T4: Insights into DNA packaging initiation and assembly of packaging motor. J. Mol. Biol. 2006, 358, 67–82. [Google Scholar] [CrossRef]
- Sun, S.; Gao, S.; Kondabagil, K.; Xiang, Y.; Rossmann, M.G.; Rao, V.B. Structure and function of the small terminase component of the DNA packaging machine in T4-like bacteriophages. Proc. Natl. Acad. Sci. USA 2012, 109, 817–822. [Google Scholar] [CrossRef]
- Büttner, C.R.; Chechik, M.; Ortiz-Lombardía, M.; Smits, C.; Ebong, I.O.; Chechik, V.; Jeschke, G.; Dykeman, E.; Benini, S.; Robinson, C.V.; et al. Structural basis for DNA recognition and loading into a viral packaging motor. Proc. Natl. Acad. Sci. USA 2012, 109, 811–816. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, L.; Rao, V.B. Exclusion of small terminase mediated DNA threading models for genome packaging in bacteriophage T4. Nucleic Acids Res. 2016, 44, 4425–4439. [Google Scholar] [CrossRef]
- Lokareddy, R.K.; Hou, C.D.; Li, F.; Yang, R.; Cingolani, G. Viral Small Terminase: A Divergent Structural Framework for a Conserved Biological Function. Viruses 2022, 14, 2215. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.G.; Wu, C.H.; Black, L.W. Reiterated gene amplifications at specific short homology sequences in phage T4 produce Hp17 mutants. J. Mol. Biol. 1991, 218, 705–721. [Google Scholar] [CrossRef]
- Black, L.W. DNA packaging and cutting by phage terminases: Control in phage T4 by a synaptic mechanism. Bioessays 1995, 17, 1025–1030. [Google Scholar] [CrossRef]
- Wu, C.H.; Lin, H.; Black, L.W. Bacteriophage T4 gene 17 amplification mutants: Evidence for initiation by the T4 terminase subunit gp16. J. Mol. Biol. 1995, 247, 523–528. [Google Scholar]
- Dixit, A.B.; Ray, K.; Black, L.W. A viral small terminase subunit (TerS) twin ring pac synapsis DNA packaging model is supported by fluorescent fusion proteins. Virology 2019, 536, 39–48. [Google Scholar] [CrossRef]
- Roy, A.; Bhardwaj, A.; Datta, P.; Lander, G.C.; Cingolani, G. Small terminase couples viral DNA binding to genome-packaging ATPase activity. Structure 2012, 20, 1403–1413. [Google Scholar] [CrossRef]
- Baumann, R.G.; Black, L.W. Isolation and characterization of T4 bacteriophage gp17 terminase, a large subunit multimer with enhanced ATPase activity. J. Biol. Chem. 2003, 278, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Black, L.W.; Peng, G. Mechanistic coupling of bacteriophage T4 DNA packaging to components of the replication-dependent late transcription machinery. J. Biol. Chem. 2006, 281, 25635–25643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondabagil, K.R.; Zhang, Z.; Rao, V.B. The DNA translocating ATPase of bacteriophage T4 packaging motor. J. Mol. Biol. 2006, 363, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Rao, V.B. The ATPase domain of the large terminase protein, gp17, from bacteriophage T4 binds DNA: Implications to the DNA packaging mechanism. J. Mol. Biol. 2008, 376, 1272–1281. [Google Scholar] [CrossRef]
- Vafabakhsh, R.; Kondabagil, K.; Earnest, T.; Lee, K.S.; Zhang, Z.; Dai, L.; Dahmen, K.A.; Rao, V.B.; Ha, T. Single-molecule packaging initiation in real time by a viral DNA packaging machine from bacteriophage T4. Proc. Natl. Acad. Sci. USA 2014, 111, 15096–15101. [Google Scholar] [CrossRef]
- Kanamaru, S.; Kondabagil, K.; Rossmann, M.G.; Rao, V.B. The functional domains of bacteriophage t4 terminase. J. Biol. Chem. 2004, 279, 40795–40801. [Google Scholar] [CrossRef]
- Rao, V.B.; Mitchell, M.S. The N-terminal ATPase site in the large terminase protein gp17 is critically required for DNA packaging in bacteriophage T4. J. Mol. Biol. 2001, 314, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.S.; Rao, V.B. Functional analysis of the bacteriophage T4 DNA-packaging ATPase motor. J. Biol. Chem. 2006, 281, 518–527. [Google Scholar] [CrossRef]
- Goetzinger, K.R.; Rao, V.B. Defining the ATPase center of bacteriophage T4 DNA packaging machine: Requirement for a catalytic glutamate residue in the large terminase protein gp17. J. Mol. Biol. 2003, 331, 139–154. [Google Scholar] [CrossRef]
- Sun, S.; Kondabagil, K.; Gentz, P.M.; Rossmann, M.G.; Rao, V.B. The structure of the ATPase that powers DNA packaging into bacteriophage T4 procapsids. Mol. Cell 2007, 25, 943–949. [Google Scholar] [CrossRef]
- Bhattacharyya, S.P.; Rao, V.B. A novel terminase activity associated with the DNA packaging protein gp17 of bacteriophage T4. Virology 1993, 196, 34–44. [Google Scholar] [CrossRef]
- Bhattacharyya, S.P.; Rao, V.B. Structural analysis of DNA cleaved in vivo by bacteriophage T4 terminase. Gene 1994, 146, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, M.; Zeng, Y.; Hu, X.; Tan, Y.; Rao, X.; Jin, X.; Li, S.; Zhu, J.; Zhang, K.; et al. Functional identification of the DNA packaging terminase from Pseudomonas aeruginosa phage PaP3. Arch. Virol. 2012, 157, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, B.J.; Hayes, J.A.; Stone, N.P.; Xu, R.G.; Kelch, B.A. The large terminase DNA packaging motor grips DNA with its ATPase domain for cleavage by the flexible nuclease domain. Nucleic Acids Res. 2017, 45, 3591–3605. [Google Scholar] [CrossRef]
- Xu, R.G.; Jenkins, H.T.; Chechik, M.; Blagova, E.V.; Lopatina, A.; Klimuk, E.; Minakhin, L.; Severinov, K.; Greive, S.J.; Antson, A.A. Viral genome packaging terminase cleaves DNA using the canonical RuvC-like two-metal catalysis mechanism. Nucleic Acids Res. 2017, 45, 3580–3590. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Cingolani, G. Structure of p22 headful packaging nuclease. J. Biol. Chem. 2012, 287, 28196–28205. [Google Scholar] [CrossRef]
- Kuebler, D.; Rao, V.B. Functional analysis of the DNA-packaging/terminase protein gp17 from bacteriophage T4. J. Mol. Biol. 1998, 281, 803–814. [Google Scholar] [CrossRef]
- Rentas, F.J.; Rao, V.B. Defining the bacteriophage T4 DNA packaging machine: Evidence for a C-terminal DNA cleavage domain in the large terminase/packaging protein gp17. J. Mol. Biol. 2003, 334, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Draper, B.; Kondabagil, K.; Rentas, F.J.; Ghosh-Kumar, M.; Sun, S.; Rossmann, M.G.; Rao, V.B. The headful packaging nuclease of bacteriophage T4. Mol. Microbiol. 2008, 69, 1180–1190. [Google Scholar] [CrossRef]
- Leffers, G.; Rao, V.B. A discontinuous headful packaging model for packaging less than headful length DNA molecules by bacteriophage T4. J. Mol. Biol. 1996, 258, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Sabanayagam, C.R.; Oram, M.; Lakowicz, J.R.; Black, L.W. Viral DNA packaging studied by fluorescence correlation spectroscopy. Biophys. J. 2007, 93, L17–L19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kottadiel, V.I.; Vafabakhsh, R.; Dai, L.; Chemla, Y.R.; Ha, T.; Rao, V.B. A promiscuous DNA packaging machine from bacteriophage T4. PLoS Biol. 2011, 9, e1000592. [Google Scholar] [CrossRef]
- Ordyan, M.; Alam, I.; Mahalingam, M.; Rao, V.B.; Smith, D.E. Nucleotide-dependent DNA gripping and an end-clamp mechanism regulate the bacteriophage T4 viral packaging motor. Nat. Commun. 2018, 9, 5434. [Google Scholar] [CrossRef] [PubMed]
- Fuller, D.N.; Raymer, D.M.; Kottadiel, V.I.; Rao, V.B.; Smith, D.E. Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proc. Natl. Acad. Sci. USA 2007, 104, 16868–16873. [Google Scholar] [CrossRef]
- Lin, S.; Alam, T.I.; Kottadiel, V.I.; VanGessel, C.J.; Tang, W.C.; Chemla, Y.R.; Rao, V.B. Altering the speed of a DNA packaging motor from bacteriophage T4. Nucleic Acids Res. 2017, 45, 11437–11448. [Google Scholar] [CrossRef] [PubMed]
- Jardine, P.J. Slow and steady wins the race: Physical limits on the rate of viral DNA packaging. Curr. Opin. Virol. 2019, 36, 32–37. [Google Scholar] [CrossRef]
- Chemla, Y.R.; Aathavan, K.; Michaelis, J.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Bustamante, C. Mechanism of force generation of a viral DNA packaging motor. Cell 2005, 122, 683–692. [Google Scholar] [CrossRef]
- Kottadiel, V.I.; Rao, V.B.; Chemla, Y.R. The dynamic pause-unpackaging state, an off-translocation recovery state of a DNA packaging motor from bacteriophage T4. Proc. Natl. Acad. Sci. USA 2012, 109, 20000–20005. [Google Scholar] [CrossRef]
- Hegde, S.; Padilla-Sanchez, V.; Draper, B.; Rao, V.B. Portal-large terminase interactions of the bacteriophage T4 DNA packaging machine implicate a molecular lever mechanism for coupling ATPase to DNA translocation. J. Virol. 2012, 86, 4046–4057. [Google Scholar] [CrossRef]
- Dai, L.; Singh, D.; Lu, S.; Kottadiel, V.I.; Vafabakhsh, R.; Mahalingam, M.; Chemla, Y.R.; Ha, T.; Rao, V.B. A viral genome packaging ring-ATPase is a flexibly coordinated pentamer. Nat. Commun. 2021, 12, 6548. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Ray, K.; Lakowicz, J.R.; Black, L.W. Dynamics of the T4 bacteriophage DNA packasome motor: Endonuclease VII resolvase release of arrested Y-DNA substrates. J. Biol. Chem. 2011, 286, 18878–18889. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W. Symmetry mismatch and DNA packaging in large bacteriophages. Proc. Natl. Acad. Sci. USA 1978, 75, 4779–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, A.A.; Tao, Y.; Leiman, P.G.; Badasso, M.O.; He, Y.; Jardine, P.J.; Olson, N.H.; Morais, M.C.; Grimes, S.; Anderson, D.L.; et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature 2000, 408, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Guasch, A.; Pous, J.; Ibarra, B.; Gomis-Ruth, F.X.; Valpuesta, J.M.; Sousa, N.; Carrascosa, J.L.; Coll, M. Detailed architecture of a DNA translocating machine: The high-resolution structure of the bacteriophage phi29 connector particle. J. Mol. Biol. 2002, 315, 663–676. [Google Scholar] [CrossRef]
- Lebedev, A.A.; Krause, M.H.; Isidro, A.L.; Vagin, A.A.; Orlova, E.V.; Turner, J.; Dodson, E.J.; Tavares, P.; Antson, A.A. Structural framework for DNA translocation via the viral portal protein. EMBO J. 2007, 26, 1984–1994. [Google Scholar] [CrossRef]
- Baumann, R.G.; Mullaney, J.; Black, L.W. Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol. Microbiol. 2006, 61, 16–32. [Google Scholar] [CrossRef]
- Hugel, T.; Michaelis, J.; Hetherington, C.L.; Jardine, P.J.; Grimes, S.; Walter, J.M.; Falk, W.; Anderson, D.L.; Bustamante, C. Experimental test of connector rotation during DNA packaging into bacteriophage phi29 capsids. PLoS Biol. 2007, 5, e59. [Google Scholar] [CrossRef]
- Draper, B.; Rao, V.B. An ATP hydrolysis sensor in the DNA packaging motor from bacteriophage T4 suggests an inchworm-type translocation mechanism. J. Mol. Biol. 2007, 369, 79–94. [Google Scholar] [CrossRef]
- Migliori, A.D.; Keller, N.; Alam, T.I.; Mahalingam, M.; Rao, V.B.; Arya, G.; Smith, D.E. Evidence for an electrostatic mechanism of force generation by the bacteriophage T4 DNA packaging motor. Nat. Commun. 2014, 5, 4173. [Google Scholar] [CrossRef]
- Migliori, A.D.; Smith, D.E.; Arya, G. Molecular interactions and residues involved in force generation in the T4 viral DNA packaging motor. J. Mol. Biol. 2014, 426, 4002–4017. [Google Scholar] [CrossRef] [PubMed]
- Oram, M.; Sabanayagam, C.; Black, L.W. Modulation of the packaging reaction of bacteriophage t4 terminase by DNA structure. J. Mol. Biol. 2008, 381, 61–72. [Google Scholar] [CrossRef]
- Aathavan, K.; Politzer, A.T.; Kaplan, A.; Moffitt, J.R.; Chemla, Y.R.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Bustamante, C. Substrate interactions and promiscuity in a viral DNA packaging motor. Nature 2009, 461, 669–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.; Sabanayagam, C.R.; Lakowicz, J.R.; Black, L.W. DNA crunching by a viral packaging motor: Compression of a procapsid-portal stalled Y-DNA substrate. Virology 2010, 398, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Golz, S.; Kemper, B. Association of holliday-structure resolving endonuclease VII with gp20 from the packaging machine of phage T4. J. Mol. Biol. 1999, 285, 1131–1144. [Google Scholar] [CrossRef]
- Dixit, A.B.; Ray, K.; Black, L.W. Compression of the DNA substrate by a viral packaging motor is supported by removal of intercalating dye during translocation. Proc. Natl. Acad. Sci. USA 2012, 109, 20419–20424. [Google Scholar] [CrossRef]
- Black, L.W.; Yan, B.; Ray, K. The T4 TerL Prohead Packaging Motor Does Not Drive DNA Translocation by a Proposed Dehydration Mechanism. Viruses 2020, 12, 522. [Google Scholar] [CrossRef]
- Harvey, S.C. The scrunchworm hypothesis: Transitions between A-DNA and B-DNA provide the driving force for genome packaging in double-stranded DNA bacteriophages. J. Struct. Biol. 2015, 189, 1–8. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Chemla, Y.R.; Aathavan, K.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Bustamante, C. Intersubunit coordination in a homomeric ring ATPase. Nature 2009, 457, 446–450. [Google Scholar] [CrossRef]
- Woodson, M.; Pajak, J.; Mahler, B.P.; Zhao, W.; Zhang, W.; Arya, G.; White, M.A.; Jardine, P.J.; Morais, M.C. A viral genome packaging motor transitions between cyclic and helical symmetry to translocate dsDNA. Sci. Adv. 2021, 7, eabc1955. [Google Scholar] [CrossRef]
- Black, L.W.; Silverman, D.J. Model for DNA packaging into bacteriophage T4 heads. J. Virol. 1978, 28, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, V.B.; Fokine, A.; Fang, Q.; Shao, Q. Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging. Viruses 2023, 15, 527. https://doi.org/10.3390/v15020527
Rao VB, Fokine A, Fang Q, Shao Q. Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging. Viruses. 2023; 15(2):527. https://doi.org/10.3390/v15020527
Chicago/Turabian StyleRao, Venigalla B., Andrei Fokine, Qianglin Fang, and Qianqian Shao. 2023. "Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging" Viruses 15, no. 2: 527. https://doi.org/10.3390/v15020527
APA StyleRao, V. B., Fokine, A., Fang, Q., & Shao, Q. (2023). Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging. Viruses, 15(2), 527. https://doi.org/10.3390/v15020527