
From Capillary Electrophoresis to Deep Sequencing: An Improved HIV-1 Drug Resistance Assessment Solution Using In Vitro Diagnostic (IVD) Assays and Software


Abstract
1. Introduction
2. Materials and Methods
2.1. Performance Evaluation
2.2. Clinical Samples
2.3. RNA Amplification
2.4. Capillary Electrophoresis Sequencing
2.5. NGS
2.6. Selection of Variant Frequency Threshold
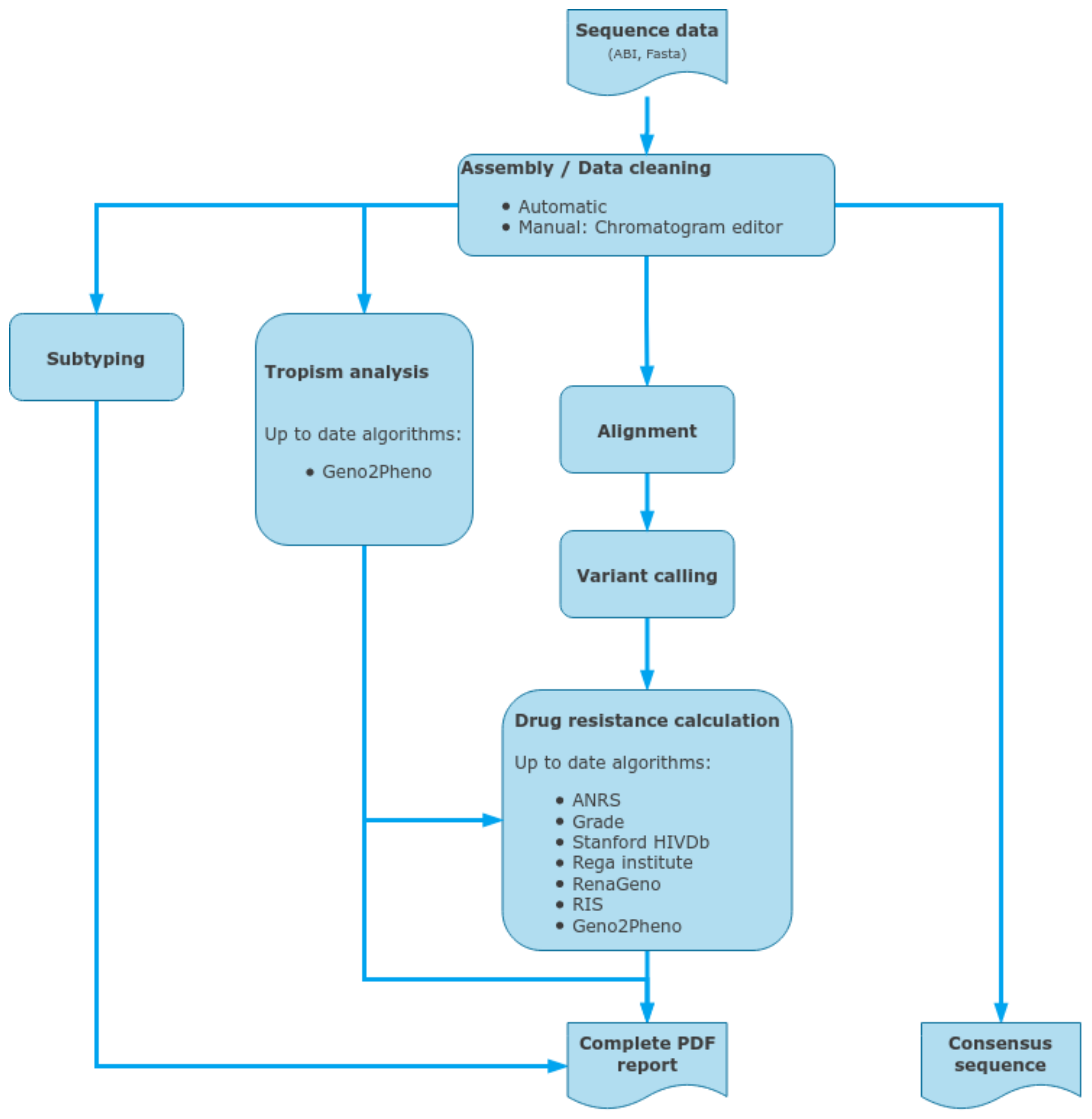
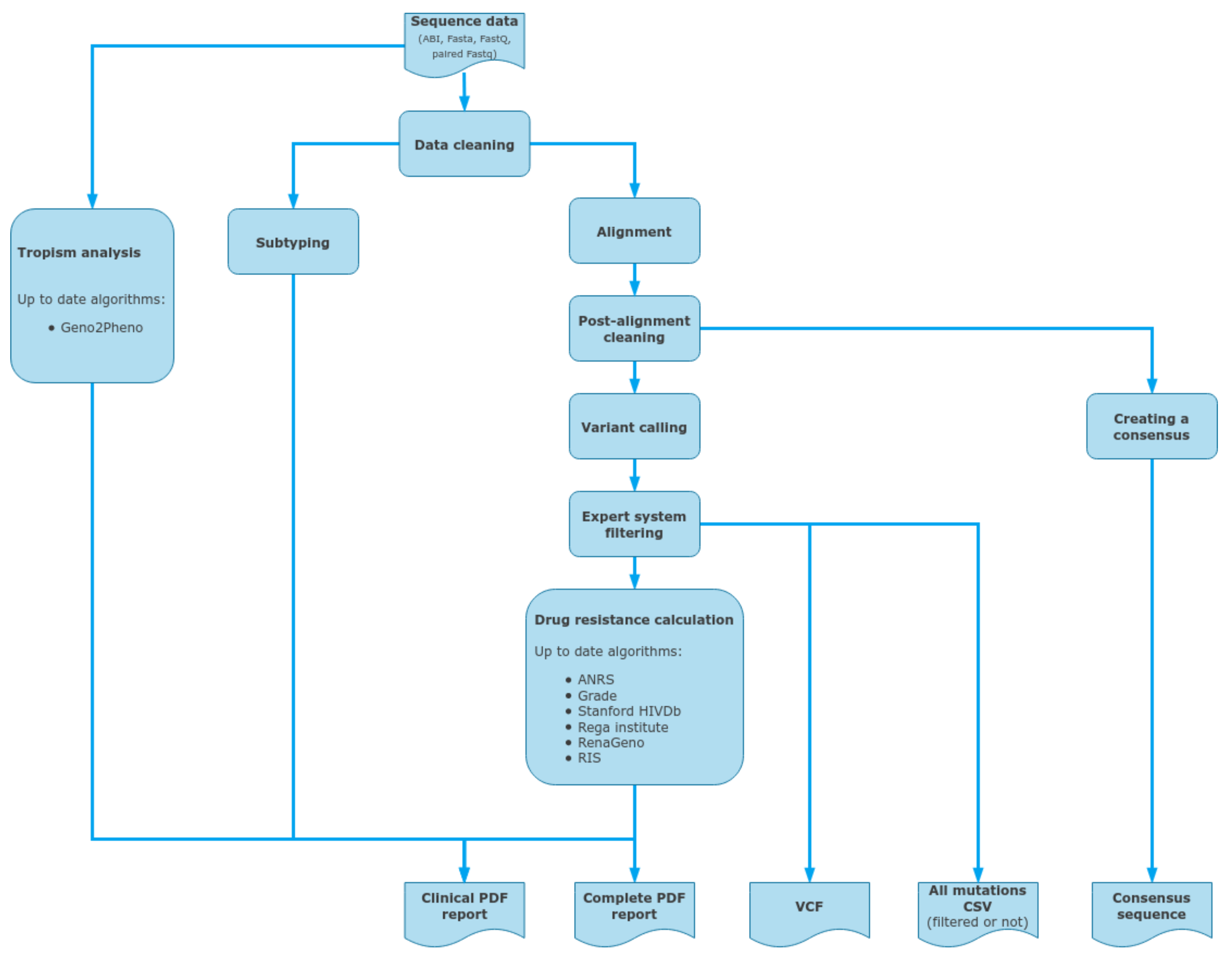
2.7. Data Analysis
2.8. ViroScore Software
2.9. DeepChek® Software
3. Results
3.1. Analytical Limit of Detection
3.2. Analytical Cutoff and Cross Reactivity
3.3. Clinical
3.4. Comparison of NGS and Capillary Electrophoresis (CE) (Sanger) Sequencing
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ávila-Ríos, S.; Parkin, N.; Swanstrom, R.; Paredes, R.; Shafer, R.; Ji, H.; Kantor, R. Next-Generation Sequencing for HIV Drug Resistance Testing: Laboratory, Clinical, and Implementation Considerations. Viruses 2020, 12, 617. [Google Scholar] [CrossRef]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadellà, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance comparison of next generation sequencing analysis pipelines for HIV-1 drug resistance testing. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Hamers, R.L.; Noguera-Julian, M.; Casadellà, M.; Parera, M.; Kityo, C.; Steegen, K.; Naniche, D.; Clotet, B.; Rinke de Wit, T.F.; et al. Clinically relevant thresholds for ultrasensitive HIV drug resistance testing: A multi-country nested case-control study. Lancet HIV 2018, 5, e638–e646. [Google Scholar] [CrossRef]
- Monaco, D.C.; Zapata, L.; Hunter, E.; Salomon, H.; Dilernia, D.A. Resistance profile of HIV-1 quasispecies in patients under treatment failure using single molecule, real-time sequencing. Aids 2020, 34, 2201–2210. [Google Scholar] [CrossRef]
- Chimukangara, B.; Giandhari, J.; Lessells, R.; Yende-Zuma, N.; Sartorius, B.; Samuel, R.; Khanyile, K.S.; Stray-Pedersen, B.; Moodley, P.; Metzner, K.J.; et al. Impact of pretreatment low-abundance HIV-1 drug-resistant variants on virological failure among HIV-1/TB-co-infected individuals. J. Antimicrob. Chemother. 2020, 75, 3319–3326. [Google Scholar] [CrossRef]
- Novitsky, V.; Nyandiko, W.; Vreeman, R.; DeLong, A.K.; Manne, A.; Scanlon, M.; Ngeresa, A.; Aluoch, J.; Sang, F.; Ashimosi, C.; et al. Added Value of Next Generation over Sanger Sequencing in Kenyan Youth with Extensive HIV-1 Drug Resistance. Microbiol. Spectr. 2022, 10. [Google Scholar] [CrossRef]
- Li, Q.; Yu, F.; Song, C.; Zhao, H.; Xiao, Q.; Lao, X.; Yang, S.; Tang, Y.; Zhang, F. HIV-1 Genotypic Resistance Testing Using Sanger and Next-Generation Sequencing in Adults with Low-Level Viremia in China. Infect. Drug Resist. 2022, ume 15, 6711–6722. [Google Scholar] [CrossRef]
- Parikh, U.M.; McCormick, K.; van Zyl, G.; Mellors, J.W. Future technologies for monitoring HIV drug resistance and cure. Curr. Opin. HIV AIDS 2017, 12, 182–189. [Google Scholar] [CrossRef]
- Jair, K.; McCann, C.D.; Reed, H.; Castel, A.D.; Pérez-Losada, M.; Wilbourn, B.; Greenberg, A.E.; Jordan, J.A.; the DC Cohort Executive Committee. Validation of publicly-available software used in analyzing NGS data for HIV-1 drug resistance mutations and transmission networks in a Washington, DC, Cohort. PLoS ONE 2019, 14, e0214820. [Google Scholar] [CrossRef]
- Gholami, M.; Rouzbahani, N.; Samiee, S.; Tayeri, K.; Ghorban, K.; Dehkharghani, A.D.; Gholami, A.A.; Moshiri, F.; Sattari, A.; Dadmanesh, M.; et al. HIV-1 drug resistance mutations detection and HIV-1 subtype G report by using next-generation sequencing platform. Microb. Pathog. 2020, 146, 104221. [Google Scholar] [CrossRef]
- Cambou, M.C.; Landovitz, R.J. Novel Antiretroviral Agents. Curr. HIV/AIDS Rep. 2020, 17, 118–124. [Google Scholar] [CrossRef]
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Dicker, I.; Zhang, S.; Ray, N.; Beno, B.R.; Regueiro-Ren, A.; Joshi, S.; Cockett, M.; Krystal, M.; Lataillade, M. Resistance profile of the HIV-1 maturation inhibitor GSK3532795 in vitro and in a clinical study. PLoS ONE 2019, 14, e0224076, Erratum in PLoS ONE 2019, 14, e0224976. [Google Scholar] [CrossRef]
- Marcelin, A.-G.; Charpentier, C.; Jary, A.; Perrier, M.; Margot, N.; Callebaut, C.; Calvez, V.; Descamps, D. Frequency of capsid substitutions associated with GS-6207 in vitro resistance in HIV-1 from antiretroviral-naive and -experienced patients. J. Antimicrob. Chemother. 2020, 75, 1588–1590. [Google Scholar] [CrossRef]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2022, 386, 1793–1803. [Google Scholar] [CrossRef]
- Armenia, D.; Santoro, M.M.; Bellocchi, M.C.; Carioti, L.; Galli, L.; Galli, A.; Scutari, R.; Salsi, E.; Mussini, C.; Sterrantino, G.; et al. Viral resistance burden and APOBEC editing correlate with virological response in heavily treatment-experienced people living with multi-drug resistant HIV. Int. J. Antimicrob. Agents 2021, 59, 106492. [Google Scholar] [CrossRef]
- Available online: https://hivfrenchresistance.org/ (accessed on 28 December 2022).
- Available online: https://hivdb.stanford.edu/ (accessed on 28 December 2022).
- Available online: https://www.iasusa.org/resources/hiv-drug-resistance-mutations/ (accessed on 28 December 2022).
- Hart, S.A.; Vardhanabhuti, S.; Strobino, S.A.; Harrison, L.J. Impact of Changes Over Time in the Stanford University Genotypic Resistance Interpretation Algorithm. Am. J. Ther. 2018, 79, e21–e29. [Google Scholar] [CrossRef]
- Mohamed, S.; Ravet, S.; Camus, C.; Khiri, H.; Olive, D.; Halfon, P. Clinical and analytical relevance of NNRTIs minority mutations on viral failure in HIV-1 infected patients. J. Med. Virol. 2013, 86, 394–403. [Google Scholar] [CrossRef]
- Mohamed, S.; Penaranda, G.; Gonzalez, D.; Camus, C.; Khiri, H.; Boulmé, R.; Sayada, C.; Philibert, P.; Olive, D.; Halfon, P. Comparison of ultra-deep versus Sanger sequencing detection of minority mutations on the HIV-1 drug resistance interpretations after virological failure. Aids 2014, 28, 1315–1324. [Google Scholar] [CrossRef]
- Moscona, R.; Ram, D.; Wax, M.; Bucris, E.; Levy, I.; Mendelson, E.; Mor, O. Comparison between next-generation and Sanger-based sequencing for the detection of transmitted drug-resistance mutations among recently infected HIV-1 patients in Israel, 2000–2014. J. Int. AIDS Soc. 2017, 20, 21846. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Beltrán, C.; Martínez-Sanz, J.; Álvarez, M.; Olalla, J.; García-Álvarez, M.; Iribarren, J.A.; Masiá, M.; Montero, M.; García-Bujalance, S.; Blanco, J.R.; et al. The algorithm used for the interpretation of doravirine transmitted drug resistance strongly influences clinical practice and guideline recommendations. J. Antimicrob. Chemother. 2020, 75, 1294–1300. [Google Scholar] [CrossRef]
- Kelentse, N.; Moyo, S.; Choga, W.T.; Lechiile, K.; Leeme, T.B.; Lawrence, D.S.; Kasvosve, I.; Musonda, R.; Mosepele, M.; Harrison, T.S.; et al. High concordance in plasma and CSF HIV-1 drug resistance mutations despite high cases of CSF viral escape in individuals with HIV-associated cryptococcal meningitis in Botswana. J. Antimicrob. Chemother. 2022, 78, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Sarinoglu, R.C.; Sili, U.; Hasdemir, U.; Aksu, B.; Soyletir, G.; Korten, V. Diversity of HIV-1 subtypes and transmitted drug-resistance mutations among minority HIV-1 variants in a Turkish cohort. Curr. HIV Res. 2021, 20, 54–62. [Google Scholar] [CrossRef]
- El Bouzidi, K.; Datir, R.P.; Kwaghe, V.; Roy, S.; Frampton, D.; Breuer, J.; Ogbanufe, O.; Murtala-Ibrahim, F.; Charurat, M.; Dakum, P.; et al. Deep sequencing of HIV-1 reveals extensive subtype variation and drug resistance after failure of first-line antiretroviral regimens in Nigeria. J. Antimicrob. Chemother. 2021, 77, 474–482. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
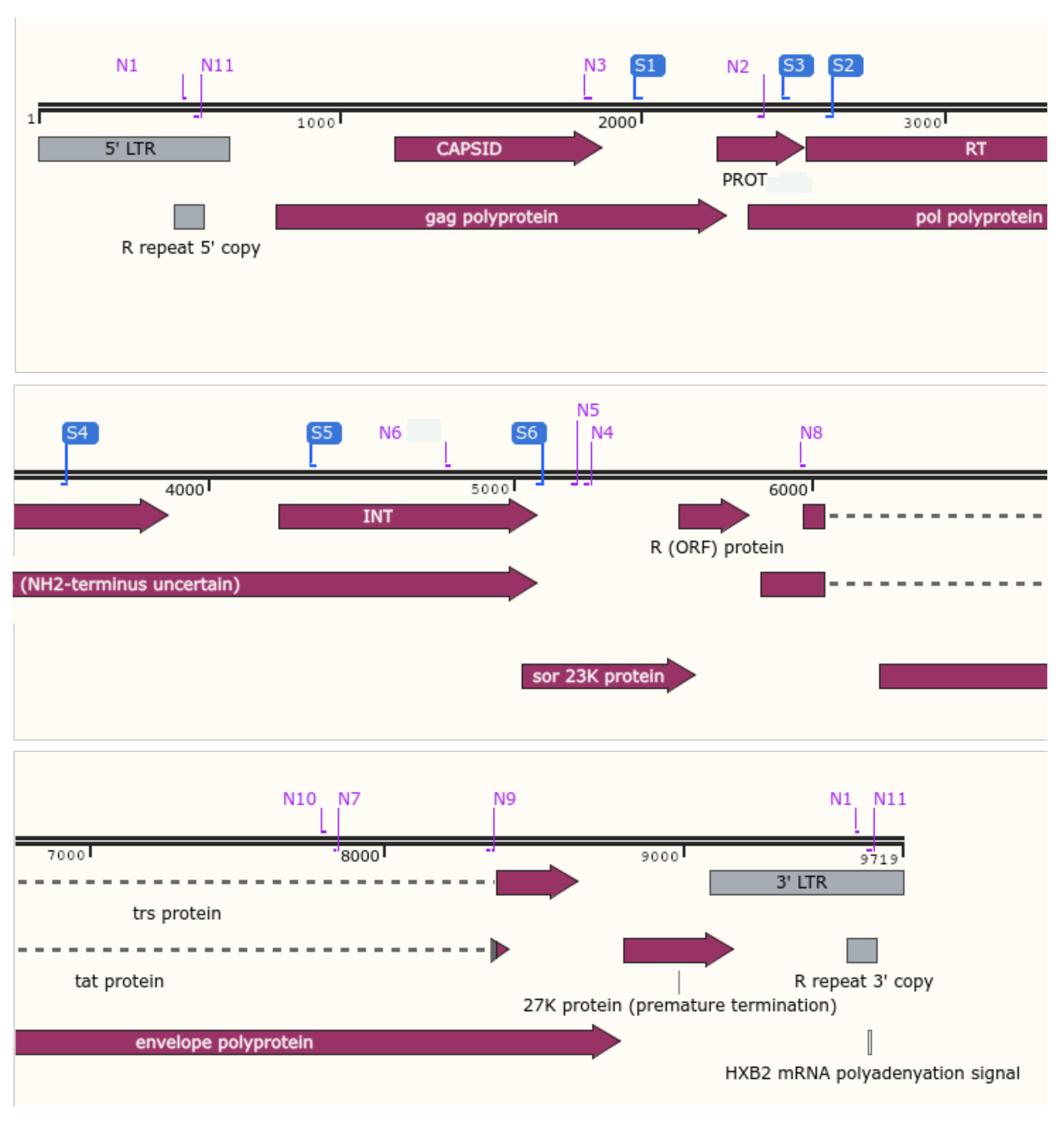
| Anti-HIV Drug Class | HIV-1 Gene Target | Target-Specific Assay (Fragment#) | Whole-Genome Assay (Fragment#) |
|---|---|---|---|
| Capsid inhibitors | gag | - | 1 |
| Nucleoside reverse transcriptase inhibitors (NRTIs) | reverse transcriptase | 1 | 2 |
| Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | reverse transcriptase | 1 | 2 |
| Protease inhibitors (PIs) | protease | 2 | 2 |
| Integrase inhibitors (IIs) | integrase | 3 | 2 |
| Integrase strand transfer inhibitors (INSTIs) | integrase | 3 | 2 |
| n.a. | vif, vpr, vpu (accessory proteins) | - | 3 |
| Fusion inhibitors | gp41 | - | 4 |
| Post-attachment inhibitors | gp120 | - | 4 |
| n.a. | nef (accessory protein) | - | 5 |
| Concentration (cp/mL) | Number of Samples Tested | Number of Correctly Identified Samples | Percentage of Correctly Identified Samples |
|---|---|---|---|
| 2000 | 13 | 13 | 100% |
| 1000 | 10 | 10 | 100% |
| 500 | 10 | 10 | 100% |
| Concentration (cp/mL) | Number of Samples Tested | Samples with Optimal Median Coverage (≥1000) | Samples with Sub-Optimal Median Coverage (>50×–< 1000) | ||
|---|---|---|---|---|---|
| Number | % | Number | % | ||
| 2000 | 13 | 13 | 100% | 0 | 0% |
| 1000 | 10 | 10 | 100% | 0 | 0% |
| 500 | 10 | 10 | 100% | 0 | 0% |
| NGS iSeq100 ANRS | NGS iSeq100 Stanford | NGS MiSeq Stanford | CE Stanford | Expected Results | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QCMD | Region | Subtype | Mutation of Interest | Subtype | Mutation of Interest | Subtype | Mutation of Interest | Subtype | Mutation of Interest | Subtype | Mutation of Interest |
| HIVDR 21S_01 | RT | C (93,67%) | M41L, E44D, D67N, T69D, A98G, M184V, L210W, T215Y | C (93,59%) | M41L, E44D, D67N, T69D, A98G, M184V, L210W, T215Y | C (93.6%) | M41L, E44D, D67N, T69D, A98G, M184V, L210W, T215Y | C (100%) | M41L, E44D, D67N, T69D, A98G, M184V, L210W, T215Y | C | M41L, E44D, D67N, T69D, A98G, M184V, L210W, T215Y |
| PR | C (92,26%) | L10F, G16E, M36V, H69K), L89M | C (92,26%) | L10F, D30N, N88D | C (92.26%) | L10F, D30N, N88D | C (100%) | L10F, D30N, N88D | C | L10F, D30N, N88D | |
| INT | C (95,71%) | ND | 08-BC (95,53%) | ND | 08_BC (95.71%) | ND | Not performed | Not performed | C | ND | |
| HIVDR 21S_02 | RT | 0206 (95,12%) | V179I | 0206 (95,12%) | ND | 0206 (95.18%) | ND | Unassigned_2;02_AG, A1 (100% similarity) | ND | AG | ND |
| PR | 02_AG (97,64%) | M36I, H69K, L89M | 02 AG (97,64%) | ND | 02_AG (97.64%) | ND | 02_AG (1) (100% similarity) | ND | AG | ND | |
| INT | 02 AG (97,65%) | ND | 02 AG (97,5%) | ND | 02_AG (97.65%) | ND | Not performed | Not performed | AG | ND | |
| HIVDR 21S_03 | RT | B (99,59%) | ND | B (99,59%) | ND | B (99.58%) | ND | B (100%similarity) | / | B | ND |
| PR | B (93,6%) | L10I, L10V, K20R, L33I, M36I, M46I, I54V, L63P, A71T, V82A, L90M | B (93,6%) | K43T (19,63%), M46I, I54V, V82A, L90M | B (93.6%) | K43T (19,69%), M46I, I54V, V82A, L90M | B (96% similarity) | M46I, I54V, V82A, L90M | B | K43T, M46I, I54V, V82A, L90M | |
| INT | B (99,45%) | ND | B (99,47%) | ND | B (99.45%) | ND | Not performed | Not performed | D | ND | |
| HIVDR 21S_04 | RT | D (95,93%) | ND | D (95,93%) | ND | D (95.81%) | ND | D (100% similarity) | ND | D | ND |
| PR | D (95,29%) | M36I, D60E, A71T | D (95,29%) | ND | D (95.29%) | ND | D (52% similarity) | ND | D | ND | |
| INT | D (97,07%) | ND | D (96,97%) | ND | D (97.07%) | ND | Not performed | Not performed | B | ND | |
| HIVDR 21S_05 | RT | C (94,49%) | M184V | B (89,2%) | M184V | B (88.81%) | M184V | C (100% similarity) | M184V | C | M184V |
| PR | C (93,6%) | G16E K20R, M36I, I54V, H69K, V82A, L89I | B (88,22%) | M46I, I54V, V82A | B (87.88%) | M46I, I54V, V82A | C (100% similarity) | M46I, I54V, V82A | C | M46I, I54V, V82A | |
| INT | B (91,27%) | ND | B (91,69%) | ND | B (91.27%) | ND | Not performed | Not performed | C | ND | |
| No. of Samples | 301 |
|---|---|
| No. of sites (median number of samples per study) | 27 (8) |
| No. of controls/EQA samples (positive/negative/EQA) | 33 (15/6/12) |
| No. of viral loads available | 215 |
| No. of viral loads ≥ 1000 cp/ml | 186 |
| Median viral load (cp/mL) | 26915 |
| No. of subtypes available | 252 |
| % of subtypes B/non-B | 63%/37% |
| No. of PR/RT or of PR/RT/INT DeepChek® Assay ran | 91/210 |
| No. of samples with viral load ≥1000 cp/mL and subtype B | 149 |
| DeepChek® Assay | Downstream Sequencing Instrument Used with DeepChek® Assay | Device 2 Used for Agreement Concordance | No. of Samples Tested | Concordance (%) |
|---|---|---|---|---|
| PR/RT + INT | Illumina MiSeq | Abbott® Dx–ViroSeq® HIV-1 Genotyping PR/RT + INT (CE) | 23 | 100% |
| PR/RT | Illumina MiSeq | LDT (German laboratory) PR/RT (CE) | 12 | 92% * |
| PR/RT + INT | Illumina MiSeq | Vela Dx–Sentosa® HIV-1 Genotyping PR/RT/INT (NGS) | 18 | 100% |
| Steps | NGS | Time/24 Samples (h) | CE | Time/24 Samples (h) |
|---|---|---|---|---|
| Sample preparation | RNA extraction kit | 1.0 | RNA extraction kit | 1.0 |
| Amplification | RT-PCR | 4 | RT-PCR | 4 |
| Purification Quantitation | Beads Purification Quality control (TapeStation) Normalization (Qubit) | 0.75 0.2 0.5 | Enzymatic purification − − | 0.2 − − |
| Library/sequencing reaction | Library preparation | 4 | Sequencing reaction | 2.5 |
| Dilution Sequencing | Dilution and pooling Sequencing | 20 | Sequencing with SeqStudio 4-capillary | 72 |
| Data analysis | FastQ files DeepChek® using ANRS, HIVdb, etc. | 0.2 | ABI files DeepChek® using ANRS, HIVdb, etc. | 1.0 |
| Result | Handling time Waiting time Time to result | 4 27 31 | Handling time Waiting time Time to result | 2 81 83 |
| Price | Reagent cost $/sample | 100–150 * | Reagent cost/sample | 80 |
| Sensitivity | 1 to 3% | 20% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, S.; Boulmé, R.; Sayada, C. From Capillary Electrophoresis to Deep Sequencing: An Improved HIV-1 Drug Resistance Assessment Solution Using In Vitro Diagnostic (IVD) Assays and Software. Viruses 2023, 15, 571. https://doi.org/10.3390/v15020571
Mohamed S, Boulmé R, Sayada C. From Capillary Electrophoresis to Deep Sequencing: An Improved HIV-1 Drug Resistance Assessment Solution Using In Vitro Diagnostic (IVD) Assays and Software. Viruses. 2023; 15(2):571. https://doi.org/10.3390/v15020571
Chicago/Turabian StyleMohamed, Sofiane, Ronan Boulmé, and Chalom Sayada. 2023. "From Capillary Electrophoresis to Deep Sequencing: An Improved HIV-1 Drug Resistance Assessment Solution Using In Vitro Diagnostic (IVD) Assays and Software" Viruses 15, no. 2: 571. https://doi.org/10.3390/v15020571
APA StyleMohamed, S., Boulmé, R., & Sayada, C. (2023). From Capillary Electrophoresis to Deep Sequencing: An Improved HIV-1 Drug Resistance Assessment Solution Using In Vitro Diagnostic (IVD) Assays and Software. Viruses, 15(2), 571. https://doi.org/10.3390/v15020571