

Dissecting Phenotype from Genotype with Clinical Isolates of SARS-CoV-2 First Wave Variants

 , , , ,
, , , ,  , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. SARS-CoV-2 Virus Isolates
2.3. Plaque Assay
2.4. Next-Generation Sequencing of NP Swabs and Virus Seed Stocks
2.5. Primer Design for UT-V1 Disjointed Tiling Amplicons
2.6. PCR Amplification and Library Preparation
2.7. Bioinformatics
2.8. In Vitro Infection of Primary Epithelial Cells
2.9. General Information and Parameters for Mouse-SARS-CoV-2 Studies
2.10. Assessment of WA1/2020 and Clinical Isolates in K18-hACE2 on 3 Days-Post-Infection
2.11. Survival Study of K18-hACE2 Mouse
2.12. Assessment of WA1/2020 and Clinical Isolates in Male and Female K18-hACE2 Mice on 1, 3, and 5 Days Post-Infection
2.13. Multiplex Immunoassay
2.14. QuantiGene Plex Assay
2.15. Histopathology and Immunohistochemistry
2.16. Neutrophil Staining and Counting
2.17. Quantification of Neutrophils via Flow Cytometry in SARS-CoV-2 Infection of K18-hACE2 Mice
2.18. Statistical Analysis
3. Results
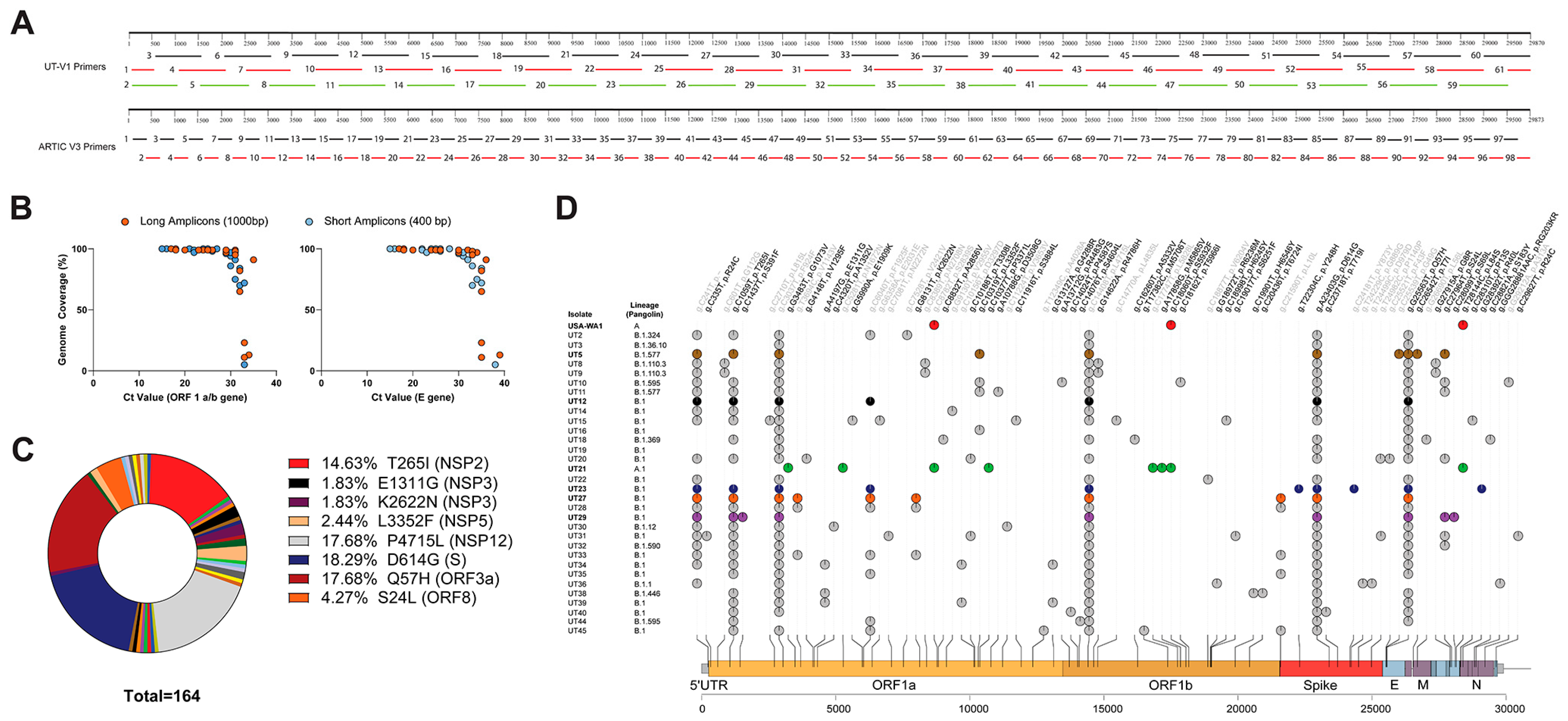
3.1. Subject Demographics, Next-Generation Sequencing, and Associated SARS-CoV-2 Lineages and SNPs
3.1.1. Subject Demographics
3.1.2. Next-Generation Sequencing
3.1.3. Bioinformatics Reveal Six Variants with Unique Constellation of Nonsynonymous Mutations
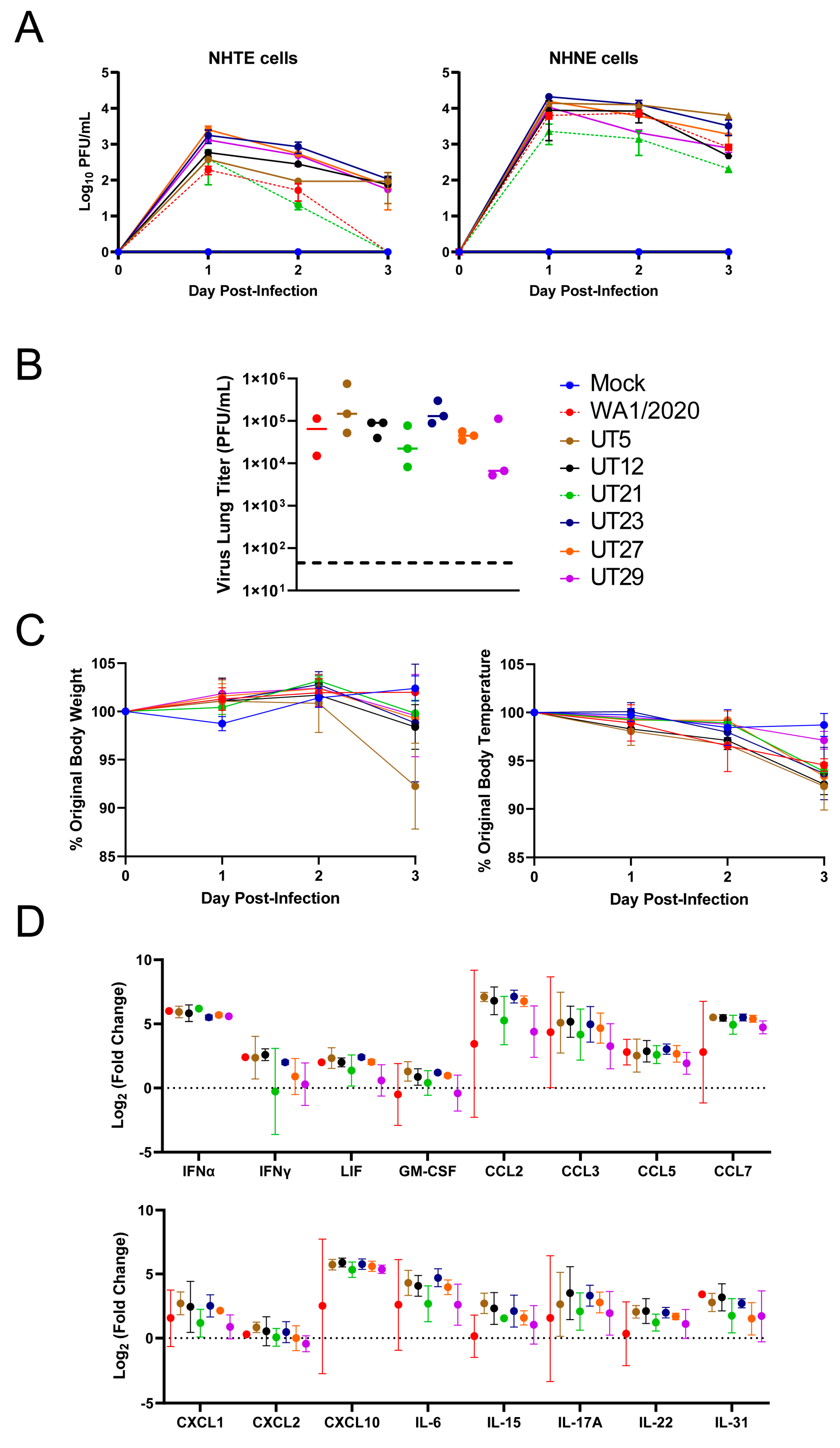
3.2. Replication Kinetics of Six Early SARS-CoV-2 Variants in Primary Epithelial Cells and Viral Load and Immune Responses in the Lungs of K18-hACE2 Mice
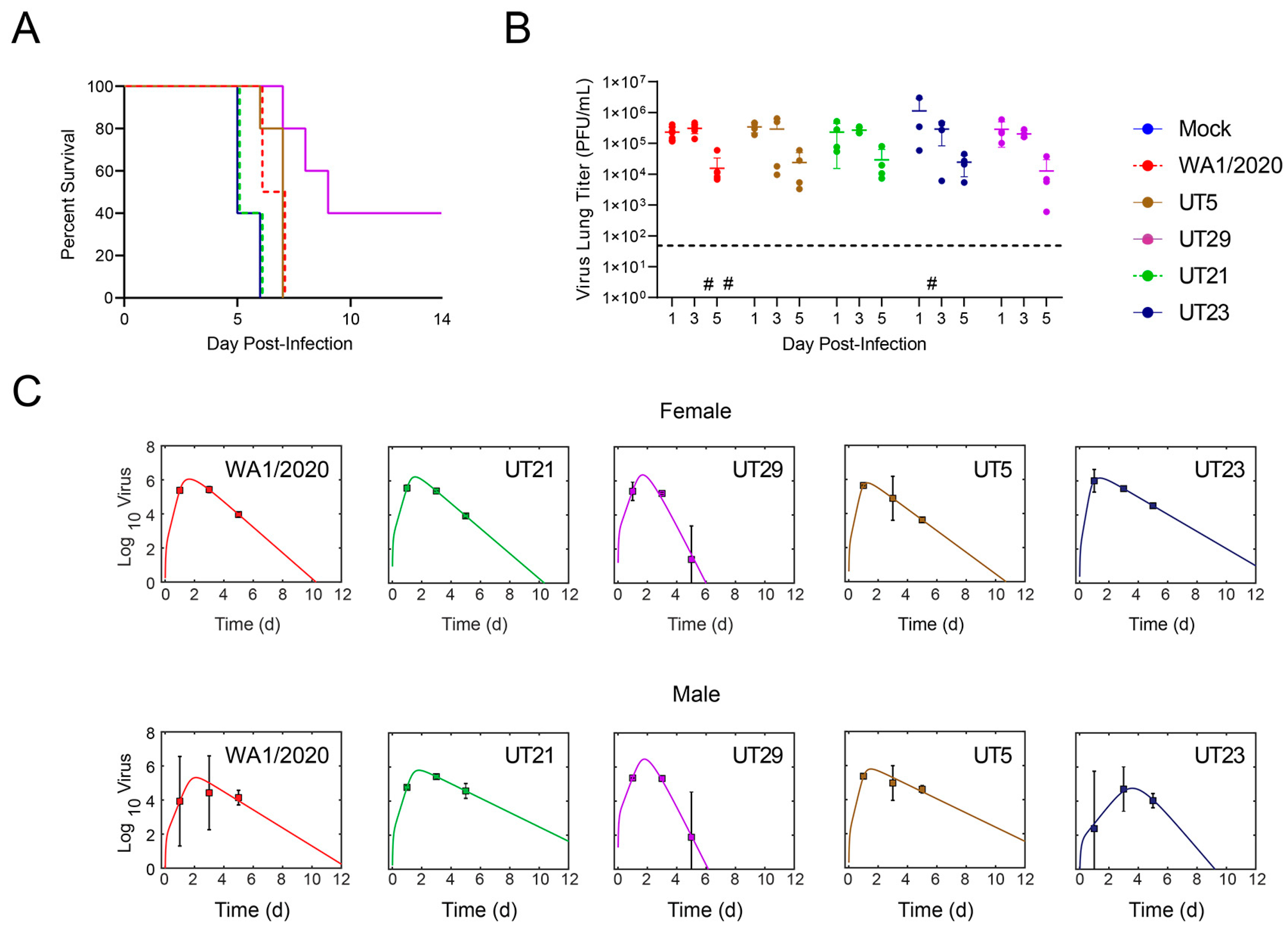
3.3. SARS-CoV-2 Clinical Isolates Yield Differences in Weight Loss and Viral Replication in the K18-hACE2 Mouse Model
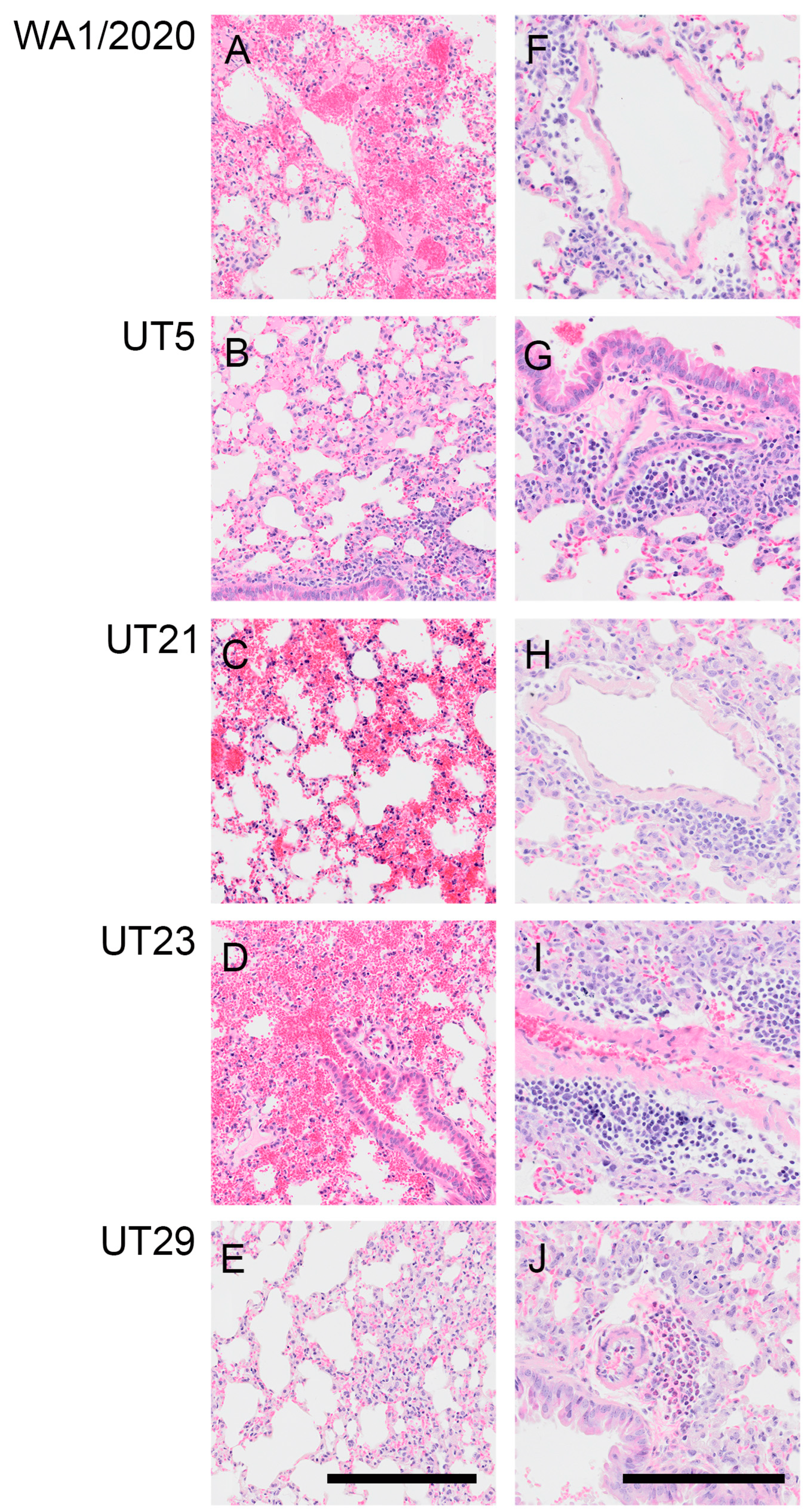
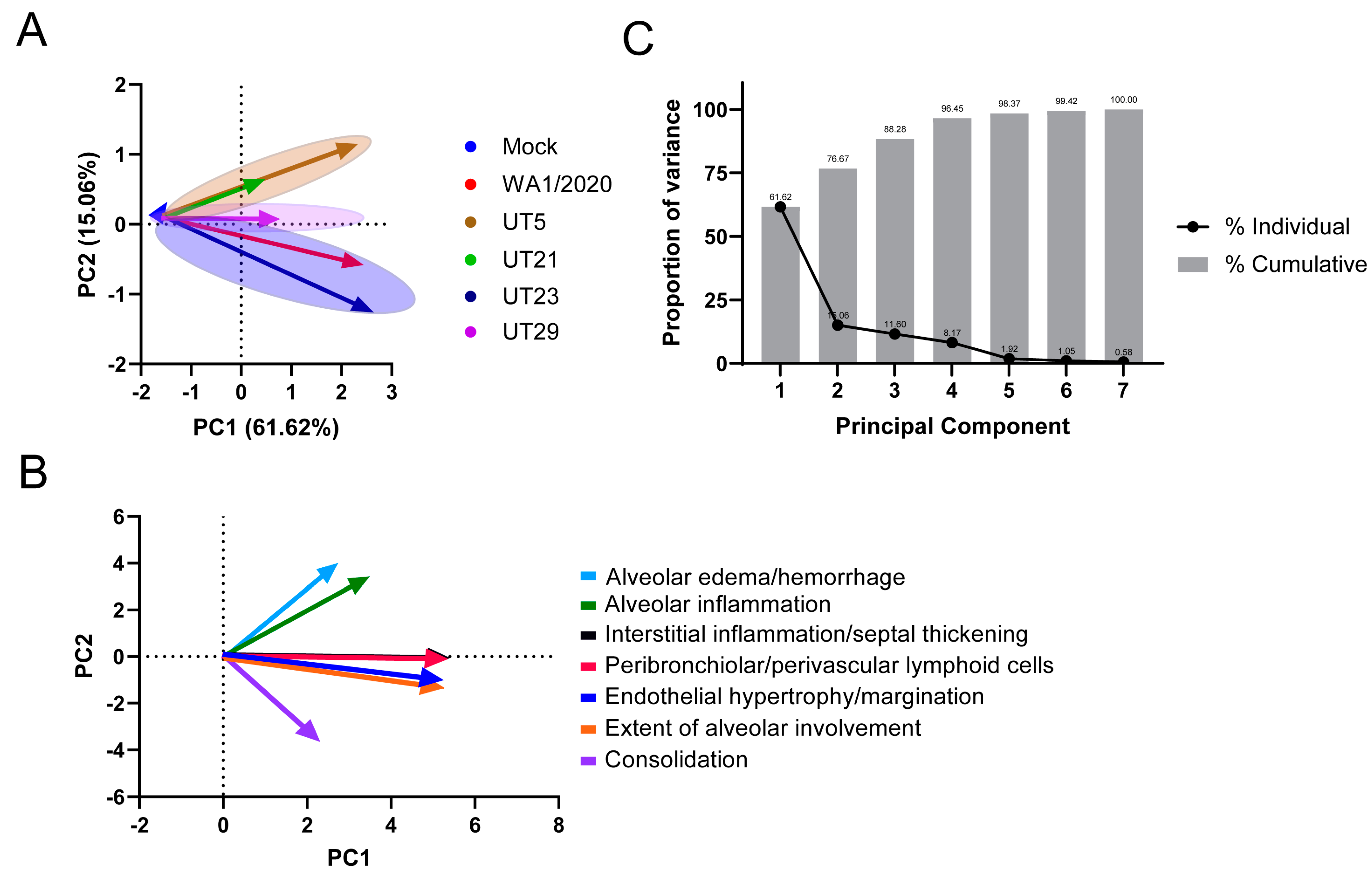
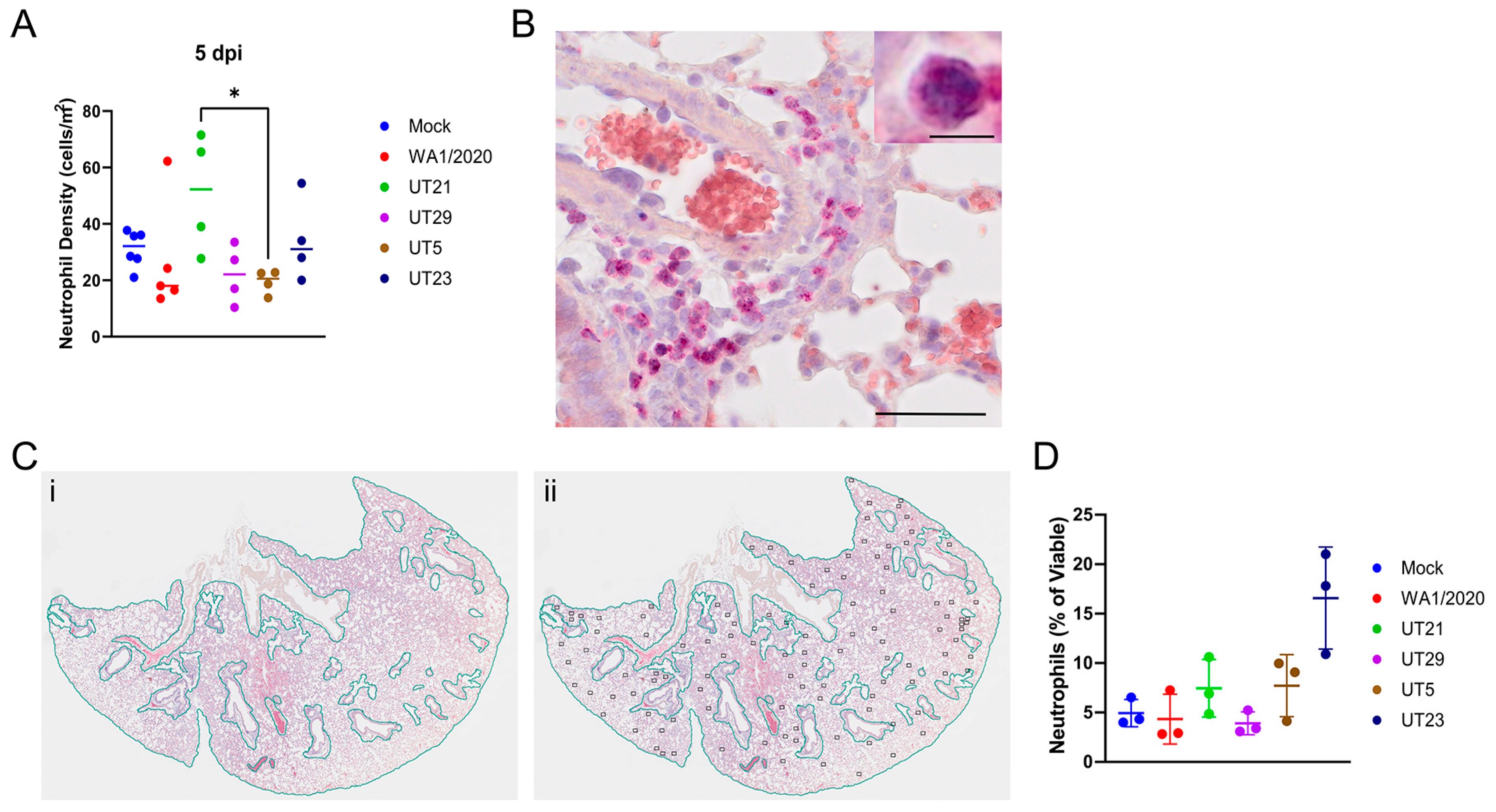
3.4. SARS-CoV-2 Variants Show Distinct Pathology Attributes in the K18-hACE2 Mouse Model
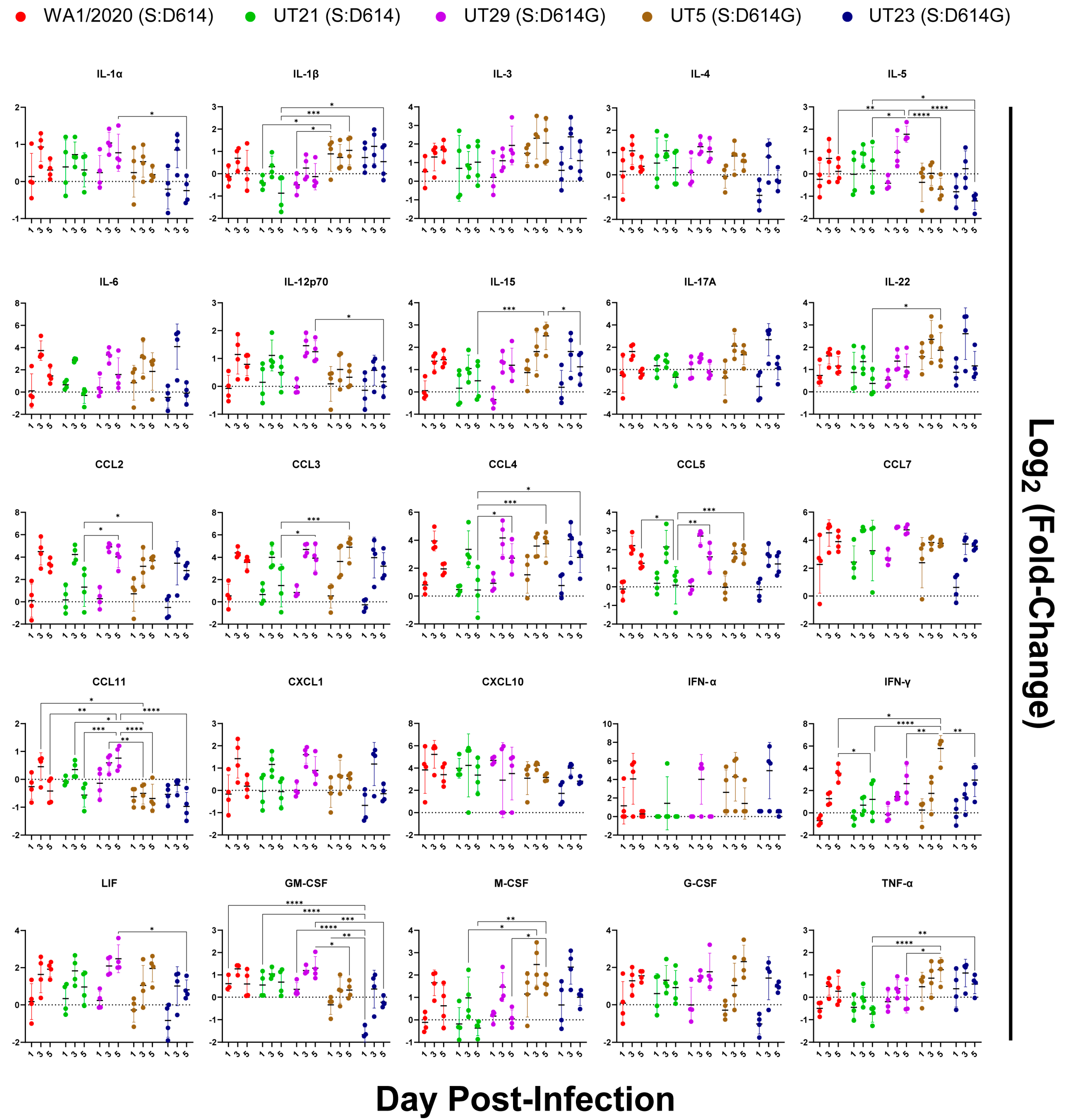
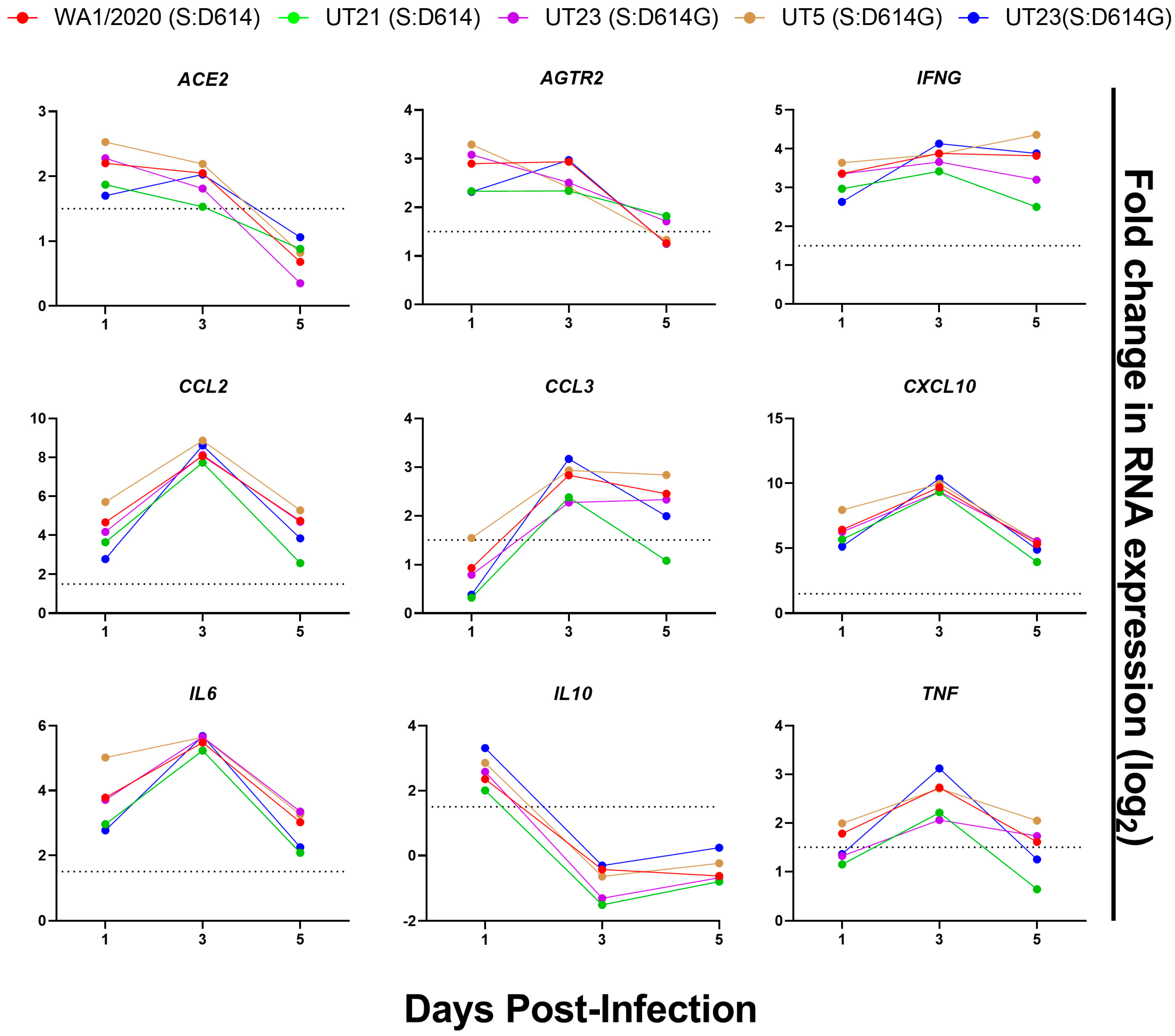
3.5. Distinct Immune Response Profiles of SARS-CoV-2 Variants in K18-hACE2 Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.J.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khan, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020, 369, 297. [Google Scholar] [CrossRef] [PubMed]
- Fauver, J.R.; Petrone, M.E.; Hodcroft, E.B.; Shioda, K.; Ehrlich, H.Y.; Watts, A.G.; Vogels, C.B.F.; Brito, A.F.; Alpert, T.; Muyombwe, A.; et al. Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell 2020, 181, 990–996.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Com. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- MacLean, O.A.; Lytras, S.; Weaver, S.; Singer, J.B.; Boni, M.F.; Lemey, P.; Kosakovsky Pond, S.L.; Robertson, D.L. Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol. 2021, 19, e3001115. [Google Scholar] [CrossRef]
- Pybus, O.G.; Rambaut, A.; Belshaw, R.; Freckleton, R.P.; Drummond, A.J.; Holmes, E.C. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 2007, 24, 845–852. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. New Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. J. 2020, 26, 1266. [Google Scholar] [CrossRef]
- Patel, D.R.; Field, C.J.; Septer, K.M.; Sim, D.G.; Jones, M.J.; Heinly, T.A.; Vanderford, T.H.; McGraw, E.A.; Sutton, T.C.; Gallagher, T. Transmission and Protection against Reinfection in the Ferret Model with the SARS-CoV-2 USA-WA1/2020 Reference Isolate. J. Virol. 2021, 95, e02232-20. [Google Scholar] [CrossRef]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., 3rd; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Zheng, J.; Wong, L.R.; Li, K.; Verma, A.K.; Ortiz, M.E.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B., Jr.; et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 2021, 589, 603–607. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; von der Thüsen, J.H.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef]
- Pannone, G.; Caponio, V.C.A.; De Stefano, I.S.; Ramunno, M.A.; Meccariello, M.; Agostinone, A.; Pedicillo, M.C.; Troiano, G.; Zhurakivska, K.; Cassano, T.; et al. Lung histopathological findings in COVID-19 disease—A systematic review. Infect Agent Cancer 2021, 16, 34. [Google Scholar] [CrossRef]
- D’Agnillo, F.; Walters, K.-A.; Xiao, Y.; Sheng, Z.-M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhao, W.; Feng, R.; Zhang, X.; Li, X.; Zhou, Y.; Peng, L.; Li, Y.; Zhang, J.; Luo, J.; et al. The pathological autopsy of coronavirus disease 2019 (COVID-2019) in China: A review. Pathog. Dis. 2020, 78, ftaa026. [Google Scholar] [CrossRef] [PubMed]
- Lowery, S.A.; Sariol, A.; Perlman, S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe 2021, 29, 1052–1062. [Google Scholar] [CrossRef]
- Oladunni, F.S.; Park, J.-G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allué-Guardia, A.; Olmo-Fontánez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Ge, Y.; Wu, B.; Zhang, W.; Wu, T.; Wen, T.; Liu, J.; Guo, X.; Huang, C.; Jiao, Y.; et al. Serum Cytokine and Chemokine Profile in Relation to the Severity of Coronavirus Disease 2019 in China. J. Infect. Dis. 2020, 222, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Research 2016, 45, D37–D42. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Consensus. Available online: https://www.hiv.lanl.gov/content/sequence/CONSENSUS/AmbigCon.html (accessed on 1 April 2020).
- OligoAnlyzer. Available online: https://www.idtdna.com/calc/analyzer (accessed on 1 April 2020).
- Artic-Network. Available online: https://github.com/artic-network/fieldbioinformatics?fbclid=IwAR02EyEqk6nnSLtPlNy3oxLUEanXzGaI8pGX-hehQC2fgk8DZq9b_NA6z6E) (accessed on 1 June 2020).
- Snippy_v4.4.0. Available online: https://github.com/tseemann/snippy (accessed on 1 June 2020).
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads. 2015. Available online: http://www.combio-lezhang.online/brucella/software_page/snippy (accessed on 1 June 2020).
- NextClade. Available online: https://clades.nextstrain.org (accessed on 15 December 2021).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Pangolin. Available online: https://github.com/cov-lineages/pangolin (accessed on 15 December 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Camp, J.V.; Bagci, U.; Chu, Y.-K.; Squier, B.; Fraig, M.; Uriarte, S.M.; Guo, H.; Mollura, D.J.; Jonsson, C.B.; Lyles, D.S. Lower Respiratory Tract Infection of the Ferret by 2009 H1N1 Pandemic Influenza A Virus Triggers Biphasic, Systemic, and Local Recruitment of Neutrophils. J. Virol. 2015, 89, 8733–8748. [Google Scholar] [CrossRef]
- Taylor, M.K.; Williams, E.P.; Wongsurawat, T.; Jenjaroenpun, P.; Nookaew, I.; Jonsson, C.B. Amplicon-Based, Next-Generation Sequencing Approaches to Characterize Single Nucleotide Polymorphisms of Orthohantavirus Species. Front. Cell. Infect. Microbiol. 2020, 10, 565591. [Google Scholar] [CrossRef]
- Ou, J.; Zhu, L.J. Trackviewer: A Bioconductor package for interactive and integrative visualization of multi-omics data. Nat. Methods 2019, 16, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The coronavirus is mutating—Does it matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.O.; Busnadiego, I.; Kufner, V.; Glas, I.; Karakus, U.; Schmutz, S.; Zaheri, M.; Abela, I.; Trkola, A.; Huber, M.; et al. SARS-CoV-2 variants reveal features critical for replication in primary human cells. PLoS Biol. 2021, 19, e3001006. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef]
- Satış, H.; Özger, H.S.; Aysert Yıldız, P.; Hızel, K.; Gulbahar, Ö.; Erbaş, G.; Aygencel, G.; Guzel Tunccan, O.; Öztürk, M.A.; Dizbay, M.; et al. Prognostic value of interleukin-18 and its association with other inflammatory markers and disease severity in COVID-19. Cytokine 2021, 137, 155302. [Google Scholar] [CrossRef]
- Fehr, A.R.; Athmer, J.; Channappanavar, R.; Phillips, J.M.; Meyerholz, D.K.; Perlman, S. The nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J. Virol. 2015, 89, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2016, 7, e01721-16. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.-P.; Malet, H.; Putics, A.; Heinonen, M.; Dutartre, H.; Frangeul, A.; Gruez, A.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; et al. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J. Virol. 2006, 80, 8493–8502. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gu, W.; Federman, S.; du Plessis, L.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.-Y.; et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef]
- Banerjee, S.; Seal, S.; Dey, R.; Mondal, K.K.; Bhattacharjee, P. Mutational spectra of SARS-CoV-2 orf1ab polyprotein and signature mutations in the United States of America. J. Med. Virol. 2021, 93, 1428–1435. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Pathirana, P.N.; Nguyen, T.; Nguyen, Q.V.H.; Bhatti, A.; Nguyen, D.C.; Nguyen, D.T.; Nguyen, N.D.; Creighton, D.; Abdelrazek, M. Genomic mutations and changes in protein secondary structure and solvent accessibility of SARS-CoV-2 (COVID-19 virus). Sci. Rep. 2021, 11, 3487. [Google Scholar] [CrossRef]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2021785118. [Google Scholar] [CrossRef]
- Thomas, S. The Structure of the Membrane Protein of SARS-CoV-2 Resembles the Sugar Transporter SemiSWEET. Pathog. Immun. 2020, 5, 342–363. [Google Scholar] [CrossRef]
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. Sars-CoV-2 Envelope and Membrane Proteins: Structural Differences Linked to Virus Characteristics? BioMed Res. Int. 2020, 2020, 4389089. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Meth. 2015, 12, 7–8. [Google Scholar] [CrossRef]
- de Haan, C.A.; de Wit, M.; Kuo, L.; Montalto-Morrison, C.; Haagmans, B.L.; Weiss, S.R.; Masters, P.S.; Rottier, P.J. The glycosylation status of the murine hepatitis coronavirus M protein affects the interferogenic capacity of the virus in vitro and its ability to replicate in the liver but not the brain. Virology 2003, 312, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Path. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C.; et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 2021, 184, 2316–2331.e15. [Google Scholar] [CrossRef]
- Yan, B.; Yang, J.; Xie, Y.; Tang, X. Relationship between blood eosinophil levels and COVID-19 mortality. World Allergy Organ. J. 2021, 14, 100521. [Google Scholar] [CrossRef]
- Zhang, J.-j.; Dong, X.; Cao, Y.-y.; Yuan, Y.-d.; Yang, Y.-b.; Yan, Y.-q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef]
- Roca, E.; Ventura, L.; Zattra, C.M.; Lombardi, C. EOSINOPENIA: An early, effective and relevant COVID-19 biomarker? QJM 2021, 114, 68–69. [Google Scholar] [CrossRef]
- Huang, R.; Xie, L.; He, J.; Dong, H.; Liu, T. Association between the peripheral blood eosinophil counts and COVID-19: A meta-analysis. Medicine 2021, 100, e26047. [Google Scholar] [CrossRef]
- Eijmael, M.; Janssens, N.; le Cessie, S.; van Dooren, Y.; Koster, T.; Karim, F. Coronavirus disease 2019 and peripheral blood eosinophil counts: A retrospective study. Infection 2021, 49, 1325–1329. [Google Scholar] [CrossRef]
- Lourda, M.; Dzidic, M.; Hertwig, L.; Bergsten, H.; Palma Medina, L.M.; Sinha, I.; Kvedaraite, E.; Chen, P.; Muvva, J.R.; Gorin, J.B.; et al. High-dimensional profiling reveals phenotypic heterogeneity and disease-specific alterations of granulocytes in COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2109123118. [Google Scholar] [CrossRef]
- Liu, F.; Xu, A.; Zhang, Y.; Xuan, W.; Yan, T.; Pan, K.; Yu, W.; Zhang, J. Patients of COVID-19 may benefit from sustained Lopinavir-combined regimen and the increase of Eosinophil may predict the outcome of COVID-19 progression. Int. J. Infect. Dis. 2020, 95, 183–191. [Google Scholar] [CrossRef] [PubMed]
- LeMessurier, K.S.; Samarasinghe, A.E. Eosinophils: Nemeses of Pulmonary Pathogens? Curr. Allergy Asthma. Rep. 2019, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R., 3rd; Kuhn, P.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-I. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Hassan, S.S.; Aljabali, A.A.A.; Panda, P.K.; Ghosh, S.; Attrish, D.; Choudhury, P.P.; Seyran, M.; Pizzol, D.; Adadi, P.; Abd El-Aziz, T.M.; et al. A unique view of SARS-CoV-2 through the lens of ORF8 protein. Comput. Biol. Med. 2021, 133, 104380. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Ma, J.; Chen, Y.; Wu, W.; Chen, Z. Structure and Function of N-Terminal Zinc Finger Domain of SARS-CoV-2 NSP2. Virol. Sin. 2021, 36, 1104–1112. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Smith, A.P.; Moquin, D.J.; Bernhauerova, V.; Smith, A.M. Influenza Virus Infection Model with Density Dependence Supports Biphasic Viral Decay. Front. Microbiol. 2018, 9, 1554. [Google Scholar] [CrossRef]
- Smith, A.M.; Adler, F.R.; McAuley, J.L.; Gutenkunst, R.N.; Ribeiro, R.M.; McCullers, J.A.; Perelson, A.S. Effect of 1918 PB1-F2 Expression on Influenza A Virus Infection Kinetics. PLoS Comput. Biol. 2011, 7, e1001081. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Adler, F.R.; Ribeiro, R.M.; Gutenkunst, R.N.; McAuley, J.L.; McCullers, J.A.; Perelson, A.S. Kinetics of Coinfection with Influenza A Virus and Streptococcus pneumoniae. PLoS Path. 2013, 9, e1003238. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Adler, F.R.; Perelson, A.S. An accurate two-phase approximate solution to an acute viral infection model. J. Math. Biol. 2010, 60, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Panday, S.K.; Alexov, E. SAAFEC-SEQ: A Sequence-Based Method for Predicting the Effect of Single Point Mutations on Protein Thermodynamic Stability. Int. J. Mol. Sci. 2021, 22, 606. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Genet. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Chen, C.-W.; Lin, J.; Chu, Y.-W. iStable: Off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinform. 2013, 14 (Suppl. S2), S5. [Google Scholar] [CrossRef]
- Molecular Operating Environment 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2022.
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2011, 40, D370–D376. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 7, 310–322. [Google Scholar]
- Taherzadeh, G.; Dehzangi, A.; Golchin, M.; Zhou, Y.; Campbell, M.P. SPRINT-Gly: Predicting N- and O-linked glycosylation sites of human and mouse proteins by using sequence and predicted structural properties. Bioinformatics 2019, 35, 4140–4146. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Verba, K.; Gupta, M.; Azumaya, C.; Moritz, M.; Pourmal, S.; Diallo, A.; Merz, G.; Jang, G.; Bouhaddou, M.; Fossati, A.; et al. CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein involved in key host processes. Res. Sq. 2021. Preprint. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Nonsynonymous Mutation | WA1 | UT21 | UT12 | UT23 | UT29 | UT27 | UT5 |
|---|---|---|---|---|---|---|---|---|
| nsp2 | T85I | |||||||
| S211F | ||||||||
| nsp3 | G255V | |||||||
| K1804N | ||||||||
| nsp5 | L89F | |||||||
| P108L | ||||||||
| nsp12 | P314L | |||||||
| nsp13 | P1427L | |||||||
| Y1464C | ||||||||
| S | Y248H | |||||||
| D614G | ||||||||
| ORF3a | Q57H | |||||||
| M | T71 | |||||||
| ORF8 | S24L | |||||||
| S69L | ||||||||
| L84S | ||||||||
| N | R40L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, M.K.; Williams, E.P.; Xue, Y.; Jenjaroenpun, P.; Wongsurawat, T.; Smith, A.P.; Smith, A.M.; Parvathareddy, J.; Kong, Y.; Vogel, P.; et al. Dissecting Phenotype from Genotype with Clinical Isolates of SARS-CoV-2 First Wave Variants. Viruses 2023, 15, 611. https://doi.org/10.3390/v15030611
Taylor MK, Williams EP, Xue Y, Jenjaroenpun P, Wongsurawat T, Smith AP, Smith AM, Parvathareddy J, Kong Y, Vogel P, et al. Dissecting Phenotype from Genotype with Clinical Isolates of SARS-CoV-2 First Wave Variants. Viruses. 2023; 15(3):611. https://doi.org/10.3390/v15030611
Chicago/Turabian StyleTaylor, Mariah K., Evan P. Williams, Yi Xue, Piroon Jenjaroenpun, Thidathip Wongsurawat, Amanda P. Smith, Amber M. Smith, Jyothi Parvathareddy, Ying Kong, Peter Vogel, and et al. 2023. "Dissecting Phenotype from Genotype with Clinical Isolates of SARS-CoV-2 First Wave Variants" Viruses 15, no. 3: 611. https://doi.org/10.3390/v15030611
APA StyleTaylor, M. K., Williams, E. P., Xue, Y., Jenjaroenpun, P., Wongsurawat, T., Smith, A. P., Smith, A. M., Parvathareddy, J., Kong, Y., Vogel, P., Cao, X., Reichard, W., Spruill-Harrell, B., Samarasinghe, A. E., Nookaew, I., Fitzpatrick, E. A., Smith, M. D., Aranha, M., Smith, J. C., & Jonsson, C. B. (2023). Dissecting Phenotype from Genotype with Clinical Isolates of SARS-CoV-2 First Wave Variants. Viruses, 15(3), 611. https://doi.org/10.3390/v15030611