
Chinese Giant Salamander Iridovirus 025L Is a Viral Essential Gene
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Plasmid Construction
2.3. Dual−Luciferase Reporter Assays, Drug Inhibition Assay
2.4. Knock down CGSIV−025L
2.5. Quantitative Real−Time PCR (qPCR) Detection
2.6. CGSIV Viral Titer
2.7. Defective Virus Was Constructed by Homologous Recombination
2.8. Interaction Analysis
2.9. Statistical Analysis
3. Results
3.1. Initiation Transcription and Transcriptional Expression of CGSIV−025L Gene
3.2. Overexpression of CGSIV−025L Promoted Virus Proliferation
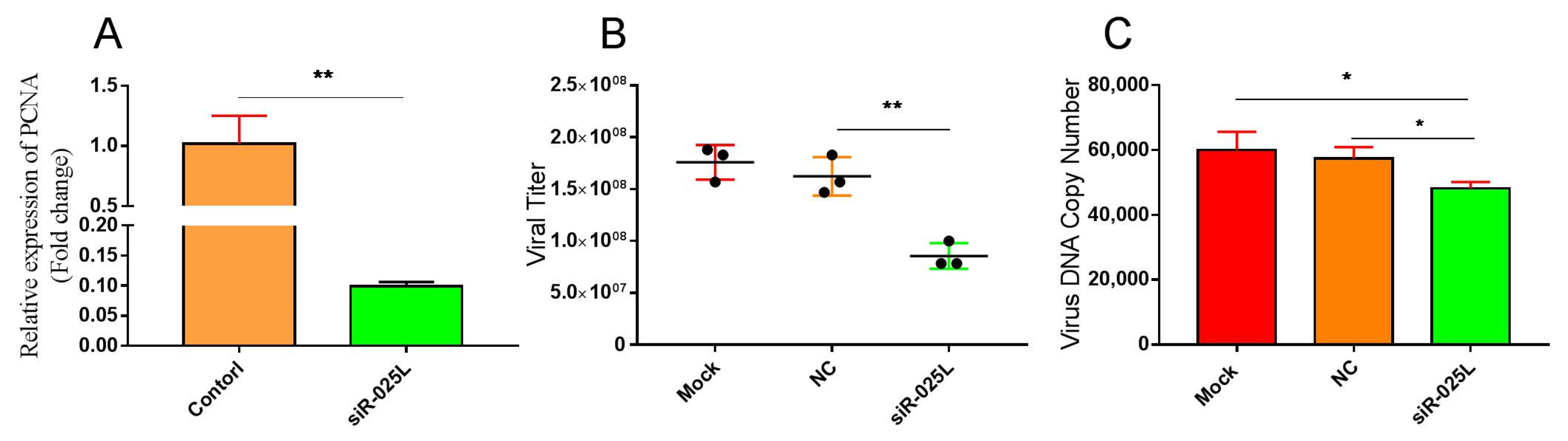
3.3. Silencing CGSIV−025L Attenuated Virus Proliferation
3.4. Deletion of CGSIV−025L Inhibited Virus Proliferation
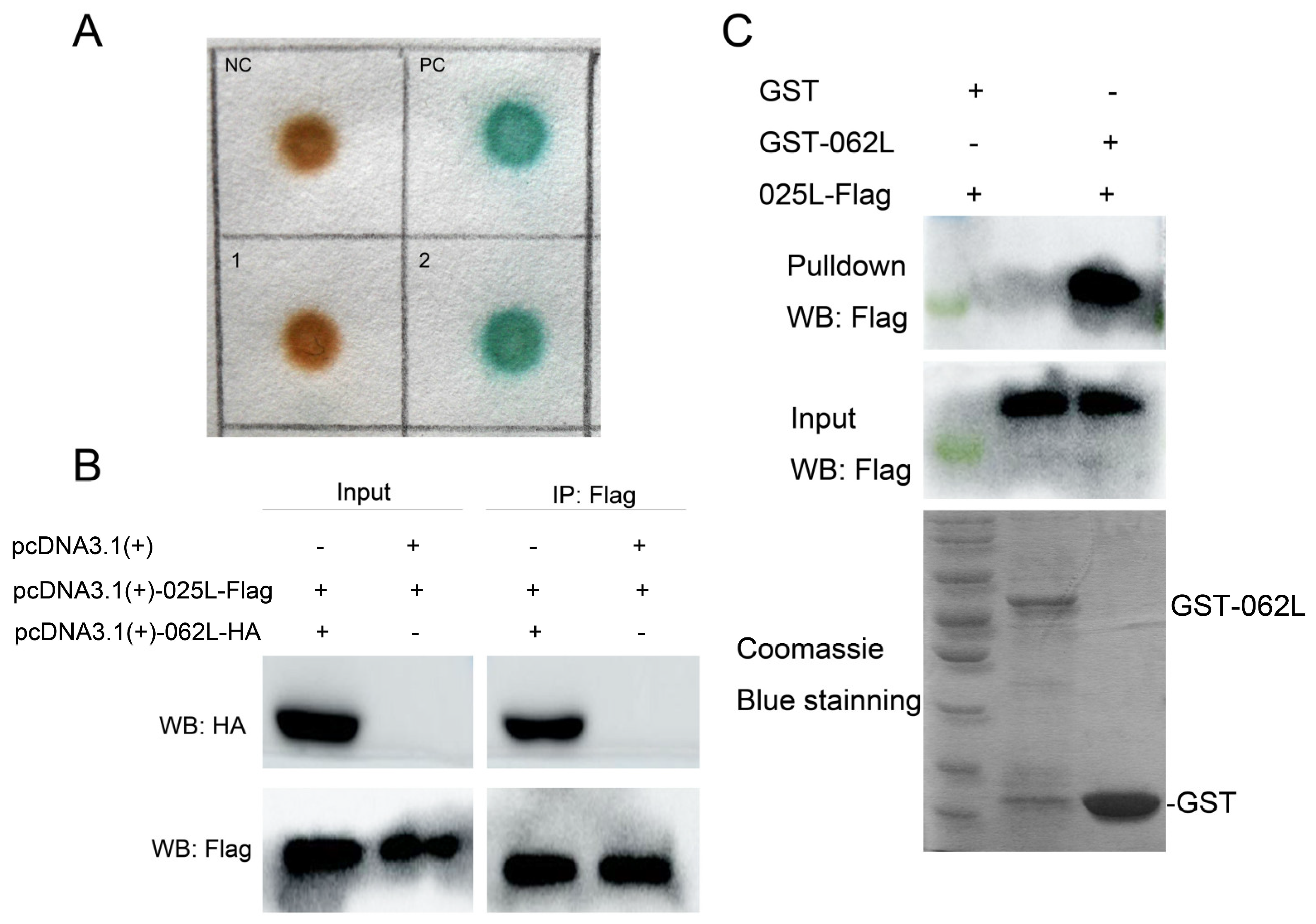
3.5. CGSIV−025L Interacts with CGSIV−062L
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ke, F.; Wang, R.; Wang, Z.; Zhang, Q. Andrias davidianus ranavirus (ADRV) genome replicate efficiently by engaging cellular mismatch repair protein MSH2. Viruses 2022, 14, 952. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, R.; de Mesquita, A.J.; Fleury, L.F.F.; de Brito, W.M.E.D.; Nunes, I.A.; Robert, J.; Morales, H.; Coelho, A.S.G.; Barthasson, D.L.; Galli, L.; et al. Mass mortality associated with a frog virus 3-like Ranavirus infection in farmed tadpoles Rana catesbeiana from Brazil. Dis. Aquat. Organ. 2009, 9, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, S.J.; Ariel, E.; Maclaine, A.; Rosa, G.M.; Gray, M.J.; Brunner, J.L.; Garner, T.W. From fish to frogs and beyond: Impact and host range of emergent ranaviruses. Virology 2017, 511, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Lin, Q.; Liu, L.; Liang, H.; Huang, Z.; Li, N. The pathogenicity and biological features of Santee-Cooper Ranaviruses isolated from Chinese perch and snakehead fish. Microb. Pathog. 2017, 112, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gui, J.; Gao, X.; Pei, C.; Hong, Y.; Zhang, Q.; Zhong, C.; Xiao, G.; Chao, P. Genome architecture changes and major gene variations of Andrias davidianus ranavirus (ADRV). Vet. Res. 2013, 44, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.; Zhang, X.; Yang, C.; An, J.; Qin, J.; Song, F.; Zeng, W. Iridovirus Infection in Chinese Giant Salamanders, China, 2010. Emerg. Infect. Dis. 2011, 17, 2388–2389. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wang, K.Y.; Zhou, Z.Y.; Li, C.W.; Wang, J.; He, M.; Yin, Z.Q.; Lai, W.M. First report of a ranavirus associated with morbidity and mortality in farmed Chinese giant salamanders (Andrias davidianus). J. Comp. Pathol. 2011, 145, 95–102. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Weng, S.; Zhao, G.; He, J.; Dong, C. Virion-associated viral proteins of a Chinese giant salamander (Andrias davidianus) iridovirus (genus Ranavirus) and functional study of the major capsid protein (MCP). Vet. Microbiol. 2014, 172, 129–139. [Google Scholar] [CrossRef]
- Ke, F.; Yu, X.-D.; Wang, Z.-H.; Gui, J.-F.; Zhang, Q.-Y. Replication and transcription machinery for ranaviruses: Components, correlation, and functional architecture. Cell Biosci. 2022, 12, 1–28. [Google Scholar] [CrossRef]
- Miyachi, K.; Fritzler, M.J.; Tan, E.M. Autoantibody to a nuclear antigen in proliferating cells. J. Immunol. 1978, 121, 2228–2234. [Google Scholar] [CrossRef]
- Mathews, M.B.; Bernstein, R.M.; Franza Jr, B.R.; Garrels, J.I. Identity of the proliferating cell nuclear antigen and cyclin. Nature 1984, 309, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Frank, R.; Blundell, P.A.; Macdonald-Bravo, H. Cyclin PCNA is the auxiliary protein of DNA polymerase-delta. Nature 1987, 326, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Prelich, G.; Tan, C.K.; Kostura, M.; Mathews, M.B.; So, A.G.; Downey, K.M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-delta aux-iliary protein. Nature 1987, 326, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Castillo, C.; So, A.G.; Downey, K.M. An auxiliary protein for DNA polymerase-delta from fetal calf thymus. J. Biol. Chem. 1986, 261, 12310–12316. [Google Scholar] [CrossRef]
- Bravo, R.; Fey, S.J.; Bellatin, J.; Larsen, P.M.; Celis, J.E. Identification of nuclear polypeptide (‘cyclin’) whose relative proportion is sensitive to changes in the rate of cell proliferation and to transformation. Prog. Clin. Biol. Res. 1982, 85, 235–248. [Google Scholar]
- Silva, L.A.; Strang, B.L.; Lin, E.W.; Kamil, J.P.; Coen, D.M. Sites and roles of phosphorylation of the human cytomegalovirus DNA polymerase subunit UL44. Virology 2011, 417, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Trego, K.S.; Zhu, Y.; Parris, D.S. The herpes simplex virus type 1 DNA polymerase processivity factor, UL42, does not alter the catalytic activity of the UL9 origin-binding protein but facilitates its loading onto DNA. Nucleic Acids Res. 2005, 33, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.I.N.; Hong, Y.U.; Dorsky, D.; Holley-Guthrie, E.; Zalani, S.; Elshiekh, N.A.; Kiehl, A.; Le, T.; Kenney, S. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1 Effects on EBV transcription and lytic replication. J. Virol. 1996, 70, 5131–5142. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ciustea, M.; Ricciardi, R.P. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology 2005, 340, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, D.; Kanda, T.; Murata, T.; Saito, S.; Sugimoto, A.; Narita, Y.; Tsurumi, T. Nuclear transport of epstein-barr virus DNA polymerase is dependent on the BMRF1 polymerase processivity factor and molecular chaperone hsp90. J. Virol. 2013, 87, 6482–6491. [Google Scholar] [CrossRef] [Green Version]
- Isomura, H.; Stinski, M.F.; Kudoh, A.; Murata, T.; Nakayama, S.; Sato, Y.; Iwahori, S.; Tsurumi, T. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 2008, 82, 1638–1646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Holley-Guthrie, E.; Ge, J.Q.; Dorsky, D.; Kenney, S. The Epstein-Barr virus (EBV) DNA polymerase accessory protein, BMRF1, activates the essential downstream component of the EBV oriLyt. Virology 1997, 230, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, H.E.; Metcalf, J.; Penny, E.; Tcherepanov, V.; Upton, C.; Brunetti, C.R. Comparative genomic analysis of the family Iridoviridae: Re-annotating and defining the core set of iridovirus genes. Virol. J. 2007, 4, 4–11. [Google Scholar] [CrossRef] [Green Version]
- He, J.G.; Lü, L.; Deng, M.; He, H.H.; Weng, S.P.; Wang, X.H.; Zhou, S.Y.; Long, Q.X.; Wang, X.Z.; Chan, S.M. Sequence analysis of the complete genome of an iridovirus isolated from the tiger frog. Virology 2002, 292, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Jancovich, J.K.; Mao, J.; Chinchar, V.G.; Wyatt, C.; Case, S.T.; Kumar, S.; Valente, G.; Subramanian, S.; Davidson, E.W.; Collins, J.P.; et al. Genomic sequence of a ranavirus (family Iridoviridae) associated with salamander mor-talities in North America. Virology 2003, 316, 90–103. [Google Scholar] [CrossRef]
- Tsai, C.T.; Ting, J.W.; Wu, M.H.; Wu, M.F.; Guo, I.C.; Chang, C.Y. Complete genome sequence of the grouper iridovirus and comparison of genomic organization with those of other iridoviruses. J. Virol. 2005, 79, 2010–2023. [Google Scholar] [CrossRef] [Green Version]
- Mavian, C.; López-Bueno, A.; Balseiro, A.; Casais, R.; Alcamí, A.; Alejo, A. The genome sequence of the emerging common midwife toad virus identifies an evolu-tionary intermediate within ranaviruses. J. Virol. 2012, 86, 3617–3625. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.G.; Barkman, T.J.; Chinchar, V.G.; Essani, K. Comparative genomic analyses of frog virus 3, type species of the genus Ranavirus (family Iridoviridae). Virology 2004, 323, 70–84. [Google Scholar] [CrossRef]
- Tidona, C.A.; Darai, G. The complete DNA sequence of lymphocystis disease virus. Virology 1997, 230, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.T.; Zhang, Q.Y. Interaction between two iridovirus core proteins and their effects on ranavirus (RGV) replication in cells from different species. Viruses 2019, 11, 416. [Google Scholar] [CrossRef] [Green Version]
- Aron, M.M.; Allen, A.G.; Kromer, M.; Galvez, H.; Vigil, B.; Jancovich, J.K. Identification of essential and non-essential genes in Ambystoma tigrinum virus. Virus Res. 2016, 217, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.; Zhang, X.; Zeng, K.; Xie, D.; Song, C.; Tam, K.; Liu, Z.; Zhou, T.; Li, W. Construction of a DNA vaccine and its protective effect on largemouth bass (Micropterus salmoides) challenged with largemouth bass virus (LMBV). Fish Shellfish Immunol. 2020, 106, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ke, F.; Zhao, Z.; Zhang, Q. Cloning, expression and subcellular distribution of a Rana grylio virus late gene encoding ERV1 homo-logue. Mol. Biol. Rep. 2008, 36, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Pallister, J.; Goldie, S.; Coupar, B.; Shiell, B.; Michalski, W.P.; Siddon, N.; Hyatt, A. Bohle iridovirus as a vector for heterologous gene expression. J. Virol. Methods 2007, 146, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Jancovich, J.K.; Jacobs, B.L. Innate immune evasion mediated by the Ambystoma tigrinum virus eukaryotic translation initiation factor 2alpha homologue. J. Virol. 2011, 85, 5061–5069. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Sheng, J.; Vu, G.P.; Liu, Y.; Foo, C.; Wu, S.; Trang, P.; Paliza-Carre, M.; Ran, Y.; Yang, X.; et al. Human cytomegalovirus UL23 inhibits transcription of interferon-γ stimulated genes and blocks antiviral interferon-γ responses by interacting with human N-myc interactor protein. PLoS Pathog. 2018, 14, e1006867. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Huang, X.; Li, S.; Sun, L.; Li, Y.; Li, H.; Zhou, Y.; Chu, Y.; Zhou, T. Identification of prosaposin as a novel interaction partner for Rhox5. J. Genet. Genom. 2007, 34, 392–399. [Google Scholar] [CrossRef]
- Abrams, A.J.; Cannatella, D.C.; Hillis, D.M.; Sawyer, S.L. Recent host-shifts in ranaviruses: Signatures of positive selection in the viral genome. J. Gen. Virol. 2013, 94, 2082–2093. [Google Scholar] [CrossRef]
- Nalçacioğlu, R.; Ince, I.A.; Vlak, J.M.; Demirbağ, Z.; van Oers, M.M. The Chilo iridescent virus DNA polymerase promoter contains an essential AAAAT motif. J. Gen. Virol. 2007, 88, 2488–2494. [Google Scholar] [CrossRef]
- Gutierrez, I.V.; Sarkar, P.; Rossetto, C.C. Kaposi’s sarcoma-associated herpesvirus processivity factor, ORF59, binds to canonical and linker histones, and its carboxy terminus is dispensable for viral DNA synthesis. J. Virol. 2021, 95, e02169-20. [Google Scholar] [CrossRef]
- Whitley, D.S.; Sample, R.C.; Sinning, A.R.; Henegar, J.; Chinchar, V.G. Antisense approaches for elucidating ranavirus gene function in an infected fish cell line. Dev. Comp. Immunol. 2011, 35, 937–948. [Google Scholar] [CrossRef]
- Sample, R.; Bryan, L.; Long, S.; Majji, S.; Hoskins, G.; Sinning, A.; Olivier, J.; Chinchar, V.G. Inhibition of iridovirus protein synthesis and virus replication by antisense morpholino oli-gonucleotides targeted to the major capsid protein, the 18 kDa immediate-early protein, and a viral homolog of RNA poly-merase II. Virology 2007, 358, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, S.; Murata, T.; Yasui, Y.; Murayama, K.; Isomura, H.; Kanda, T.; Tsurumi, T. Tetrameric ring formation of epstein-barr virus polymerase processivity factor is crucial for viral replication. J. Virol. 2010, 84, 12589–12598. [Google Scholar] [CrossRef] [Green Version]
- Javed, A.; Major, B.; Stead, J.A.; Sanders, C.M.; Orlova, E.V. Unwinding of a DNA replication fork by a hexameric viral helicase. Nat. Commun. 2021, 12, 5535. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| 025L−F1 | CCCAAGCTTATGCTGTGGGAAGCCGTAA |
| 025L−R1 | GGAATTCTGCCCTCAAAGAGAGTCACGG |
| 025L−F2 | CGGAATTCGCCACCATGCTGTGGGAAGCCGTA |
| 025L−R2 | GCGTCGACTTAGCCCTCAAAGAGAGTCACG |
| 025L−F3 | GGGGTACCCTCGTAACGACTG |
| 025L−R3 | CCCAAGCTTTGACTGGTTTATC |
| puro−F | CAGTGTGGTGGAATTATGACCGAGTACAAGCCCACG |
| puro−R | AGTAGCTCCGCTTCCGGCACCGGGCTTGCG |
| RFP−F | GGAAGCGGAGCTACTAAC |
| RFP−R | AAACGGGCCCTCTAGTCATCTGTGCCCCAGTTTG |
| 062L−F1 | CGGAATTCGCCACCATGGCAAAACTTTTAAGGC |
| 062L−R1 | CCCTCGAGCTACCTCTGCGGTCGTCG |
| 062L−F2 | CGGAATTCGCCACCATGGCAAAACTTTTAAGGC |
| 062L−R2 | CCCTCGAGTTAAGCGTAATCTGGAACATCGTATGGGTACCTCTGCGGTCGTCG |
| qICP−46−F | GGTCCTTGTTCAGATTCGC |
| qICP−46−R | AATCAGGGCTCTGGTTATG |
| qMCP−F | CTGGAGAAGAAGAATGGGAGGGG |
| qMCP−R | CTTTCGGGCAGCAGTTTTCGGTC |
| q025L−F | ATCTTGACGAGCCTGGACATCTTT |
| q025L−R | GTCCTCGGTCTTTACCAGCTACGT |
| β−actin−F | CACTGTGCCCATCTACGAG |
| β−actin−R | CCATCTCCTGCTCGAAGTC |
| q18S−F | ATGGTACTTTAGGCGCCTAC |
| q18S−R | TATACGCTATTGGAGCTGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Xie, D.; Nong, S.; Wu, Y.; Huang, S.; He, X.; Zhou, T.; Li, W. Chinese Giant Salamander Iridovirus 025L Is a Viral Essential Gene. Viruses 2023, 15, 617. https://doi.org/10.3390/v15030617
Liu Z, Xie D, Nong S, Wu Y, Huang S, He X, Zhou T, Li W. Chinese Giant Salamander Iridovirus 025L Is a Viral Essential Gene. Viruses. 2023; 15(3):617. https://doi.org/10.3390/v15030617
Chicago/Turabian StyleLiu, Zijing, Daofa Xie, Shirong Nong, Yingzi Wu, Suxian Huang, Xianhui He, Tianhong Zhou, and Wei Li. 2023. "Chinese Giant Salamander Iridovirus 025L Is a Viral Essential Gene" Viruses 15, no. 3: 617. https://doi.org/10.3390/v15030617
APA StyleLiu, Z., Xie, D., Nong, S., Wu, Y., Huang, S., He, X., Zhou, T., & Li, W. (2023). Chinese Giant Salamander Iridovirus 025L Is a Viral Essential Gene. Viruses, 15(3), 617. https://doi.org/10.3390/v15030617