
Heparan Sulfate and Enoxaparin Interact at the Interface of the Spike Protein of HCoV-229E but Not with HCoV-OC43

 , , ,
, , ,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Cellular Lines, and Viruses
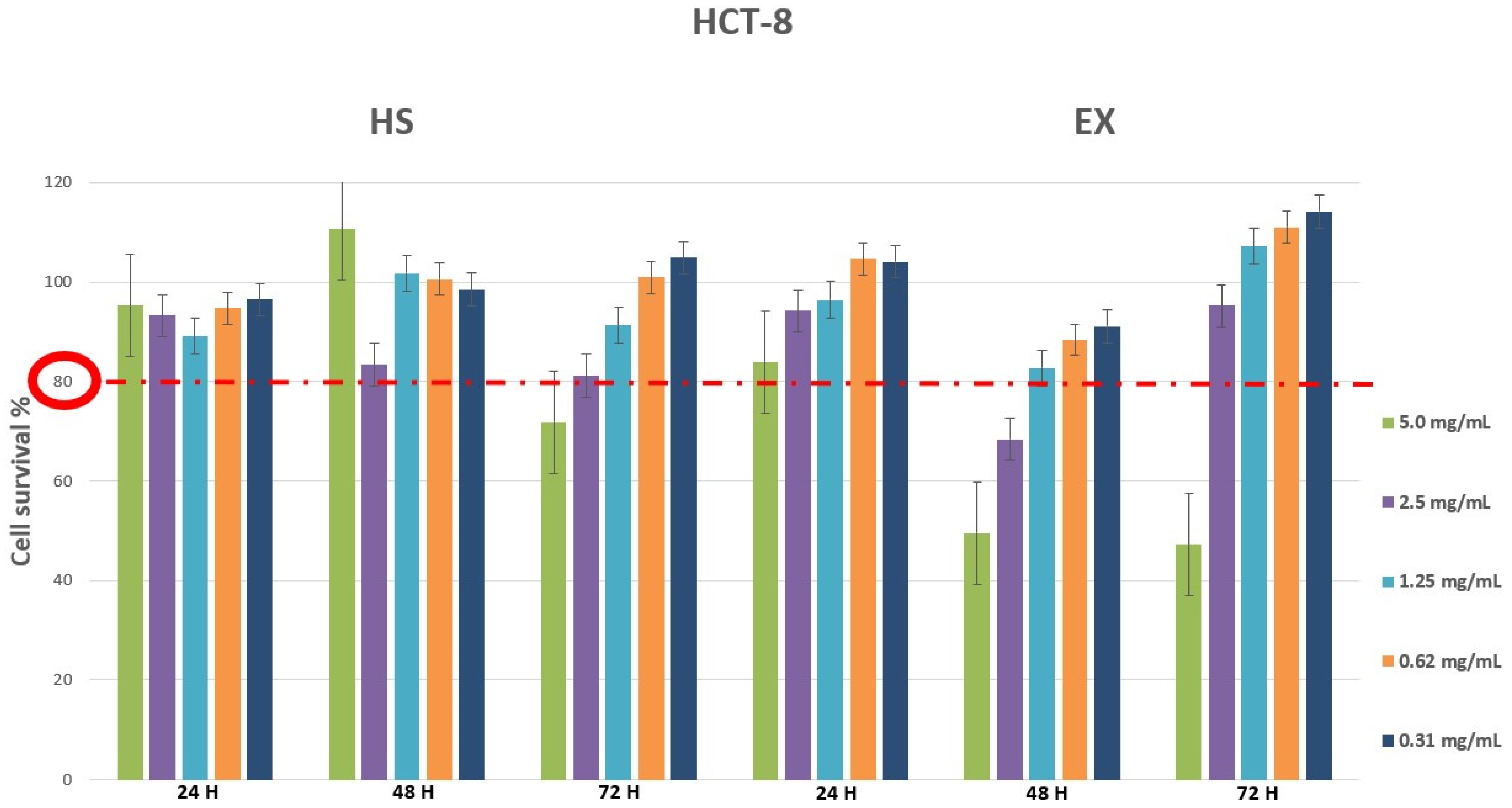
2.2. Cell Viability Assay by MTT
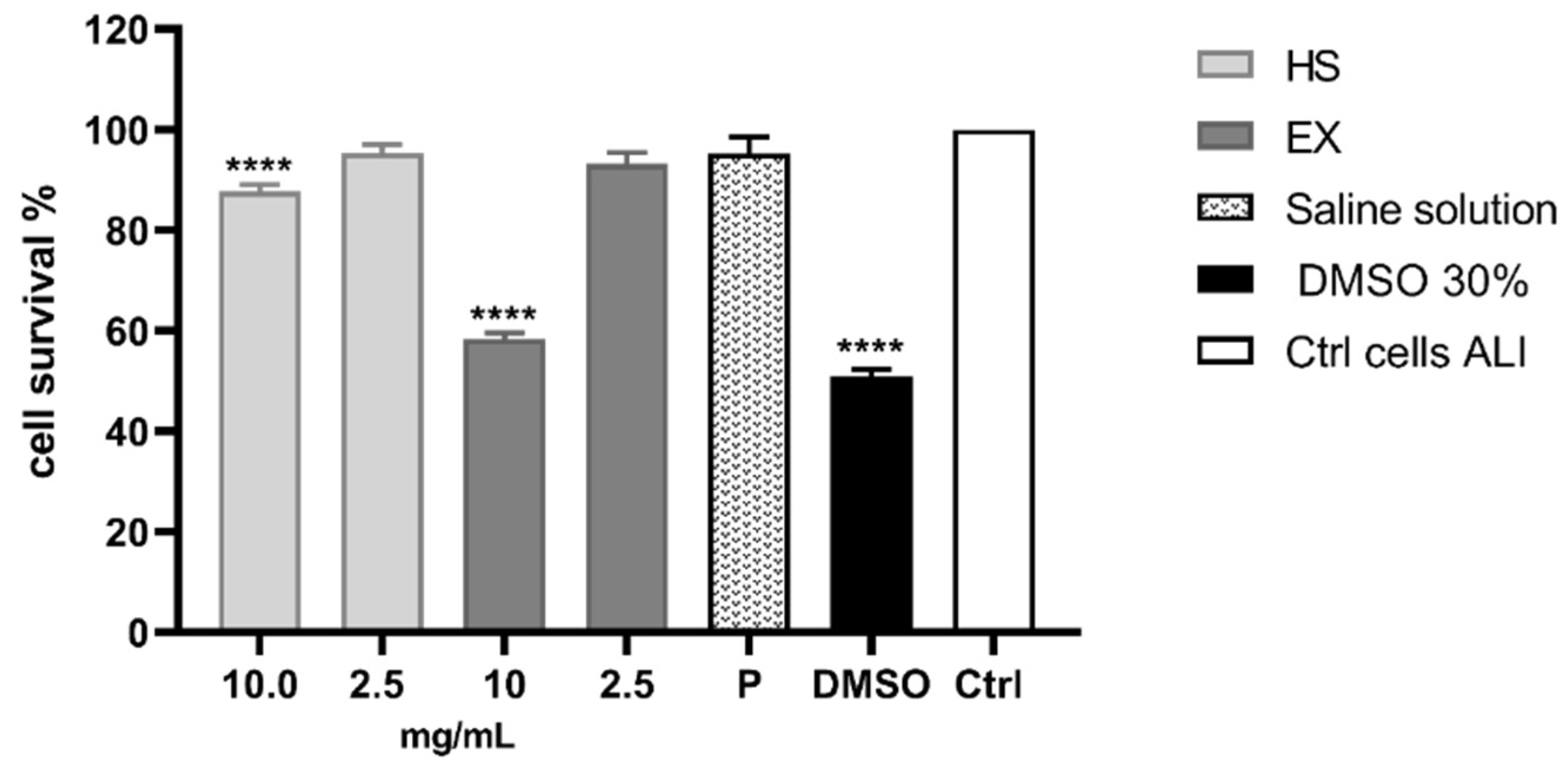
2.3. Cell Viability Assay by Air-Liquid Interface (ALI) Exposure
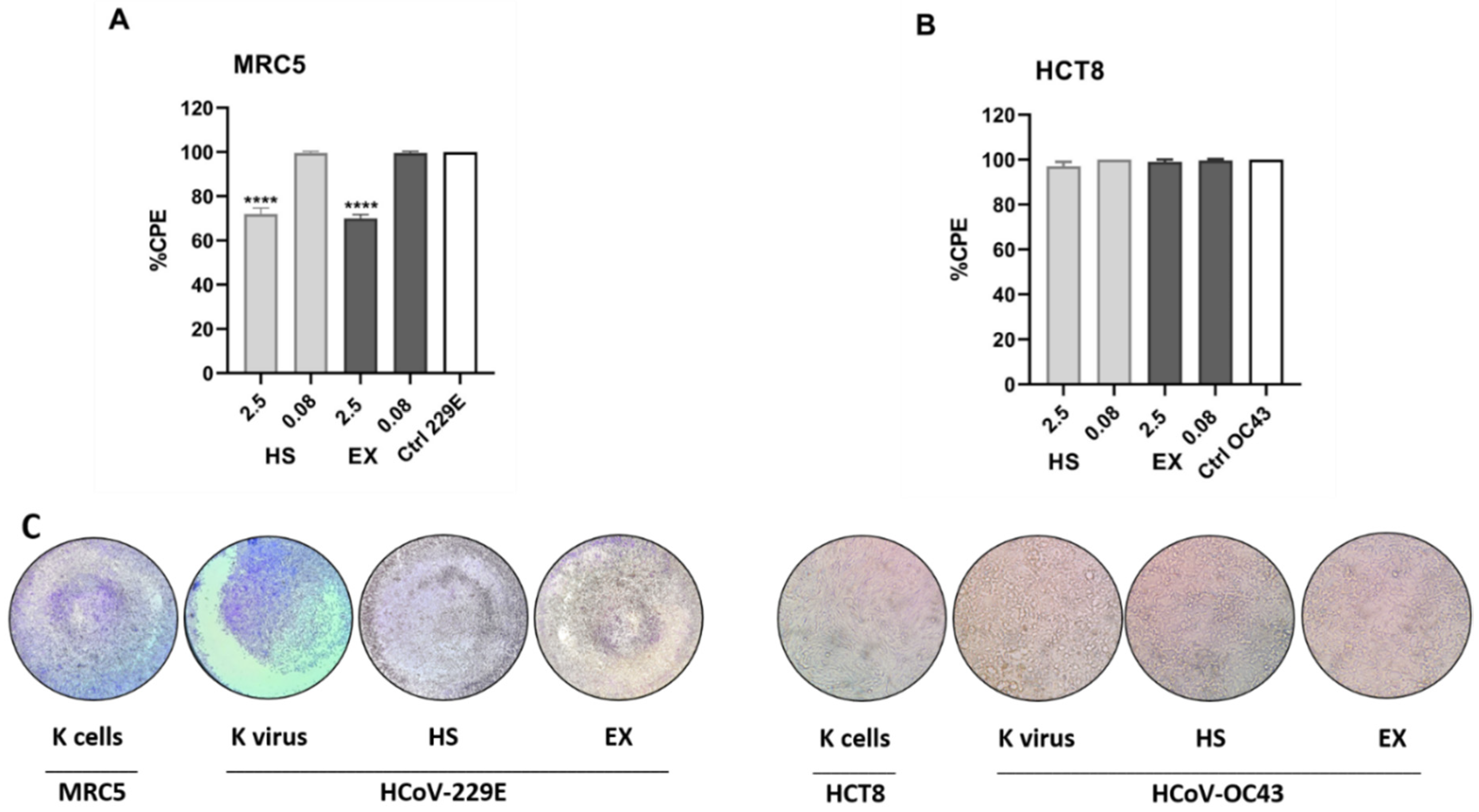
2.4. Antiviral Activity
2.5. Statistical Analysis
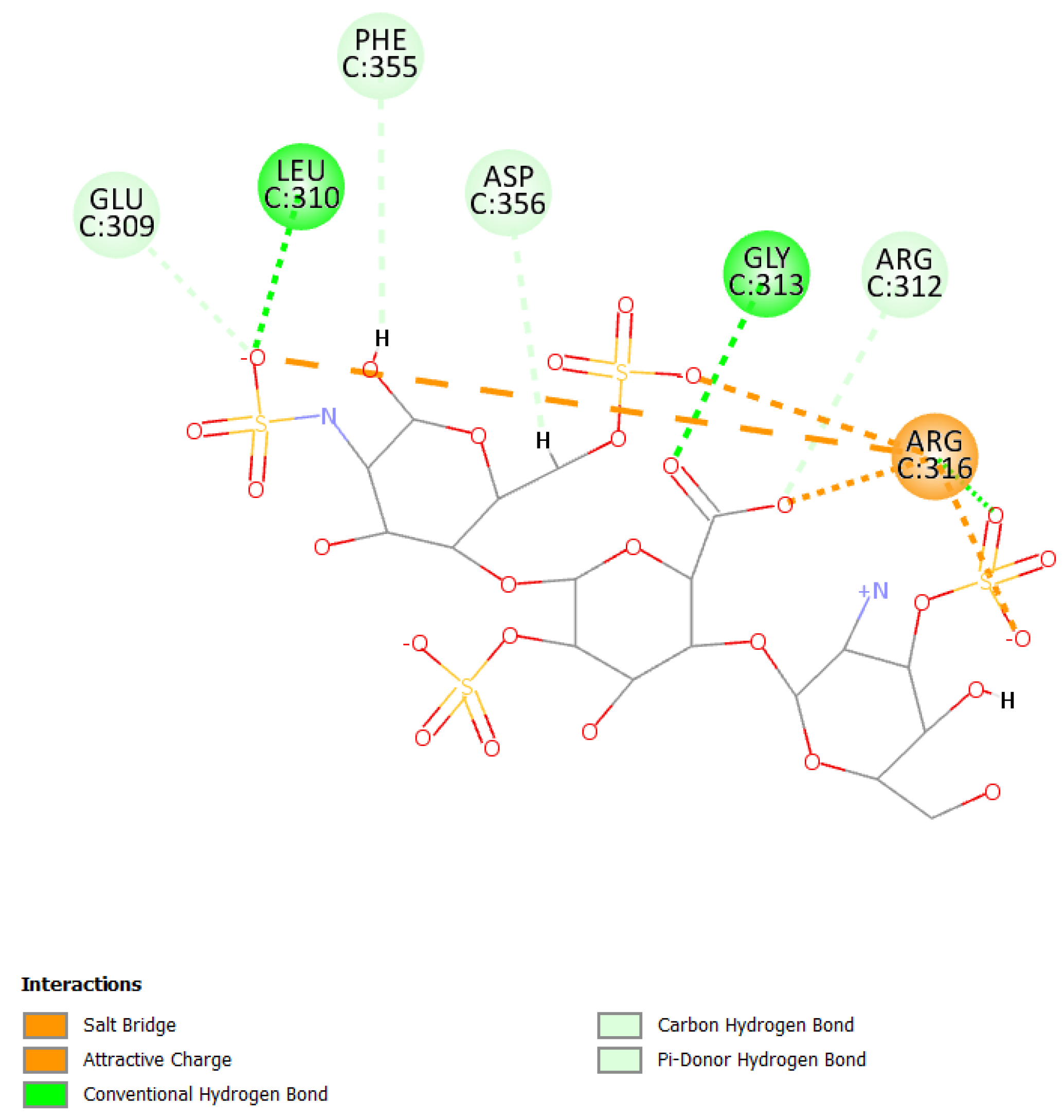
2.6. Molecular Modeling
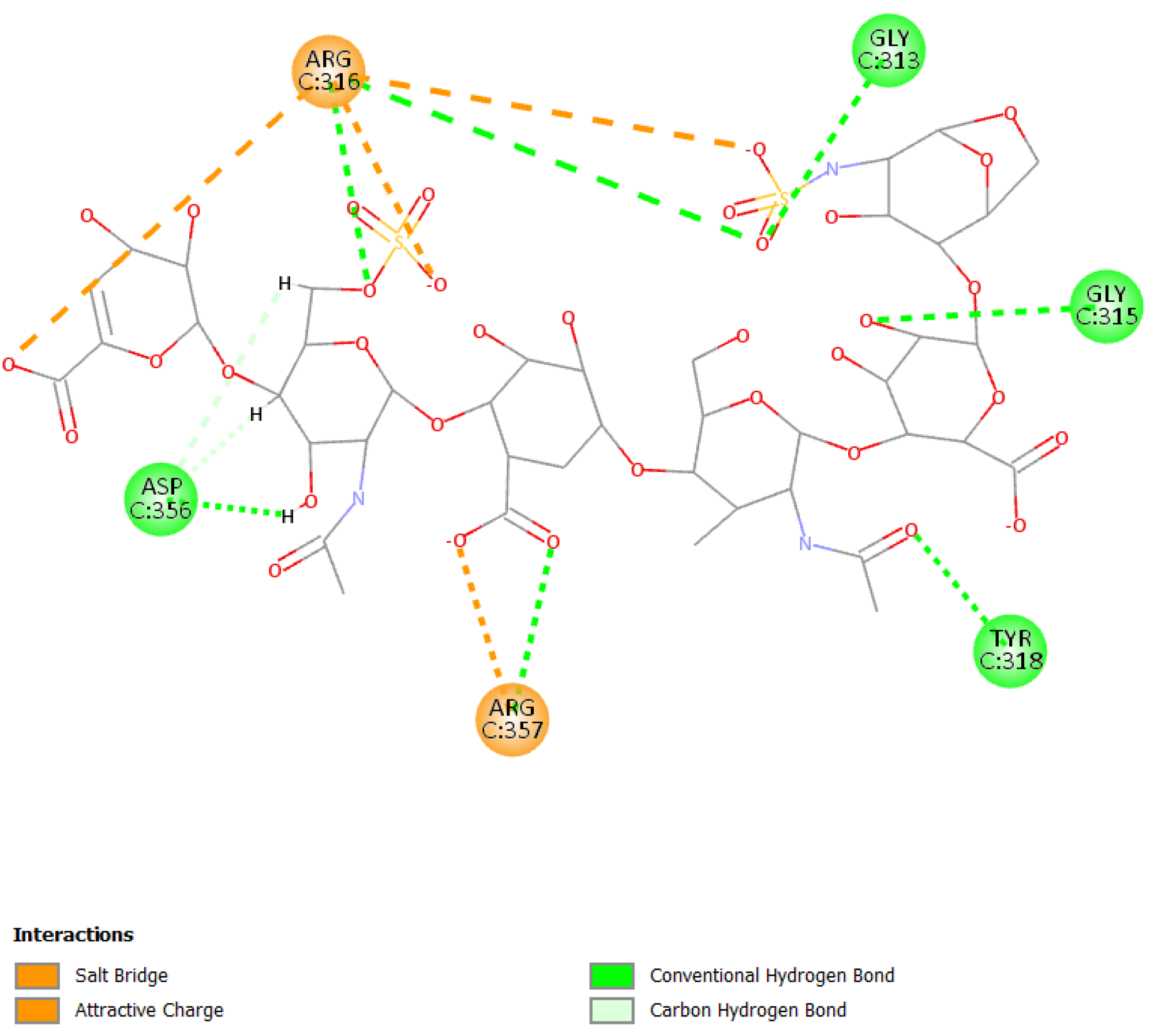
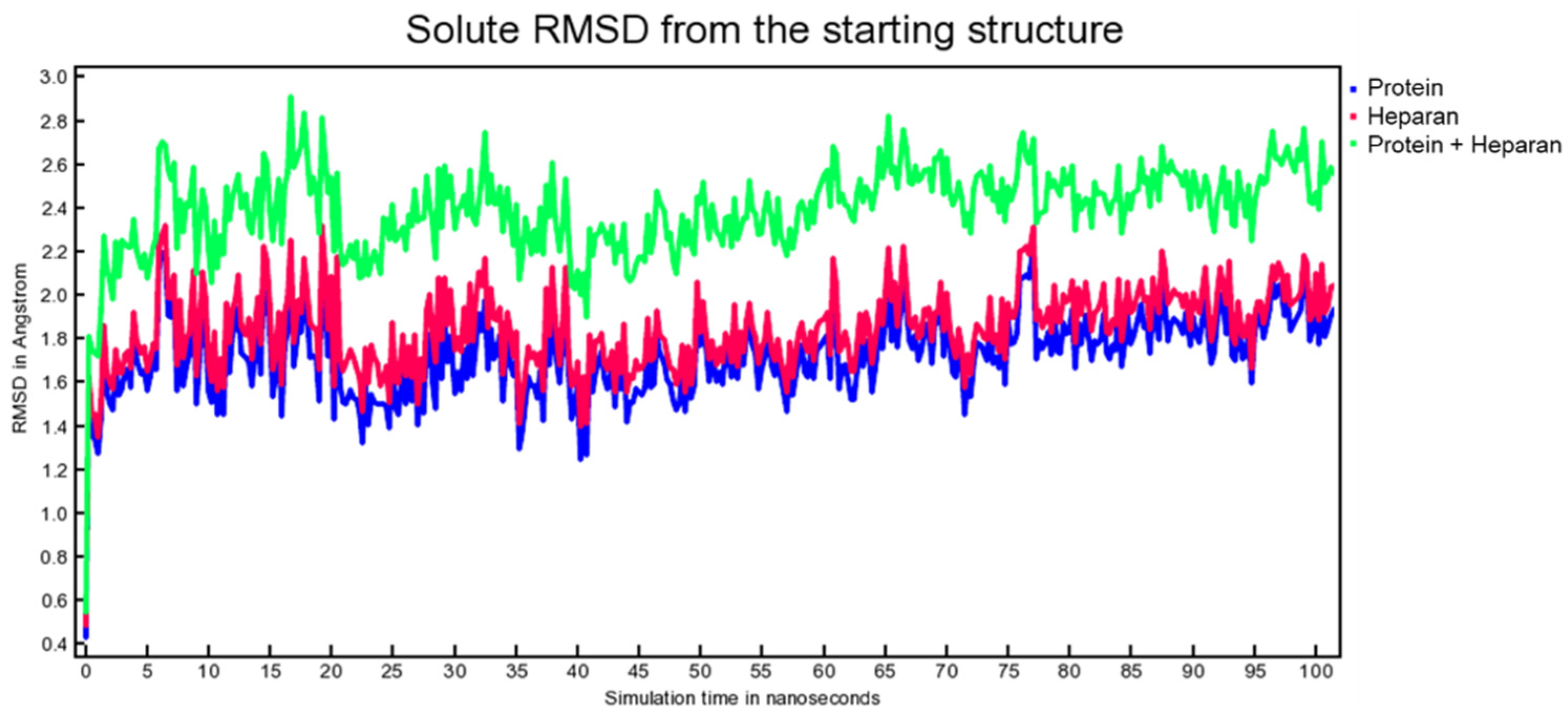
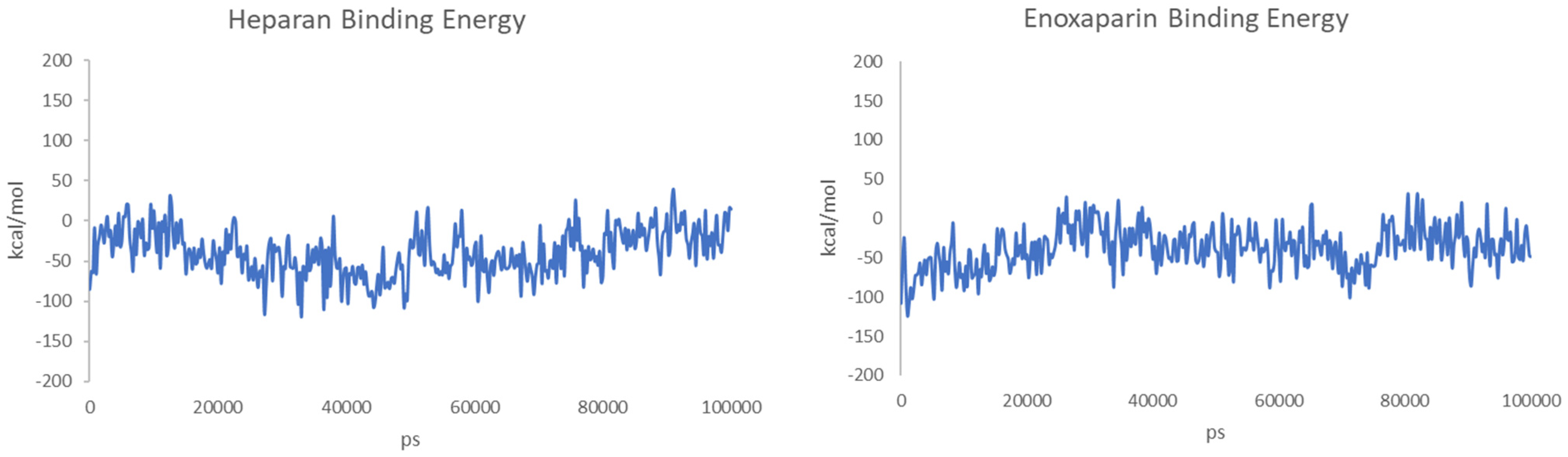
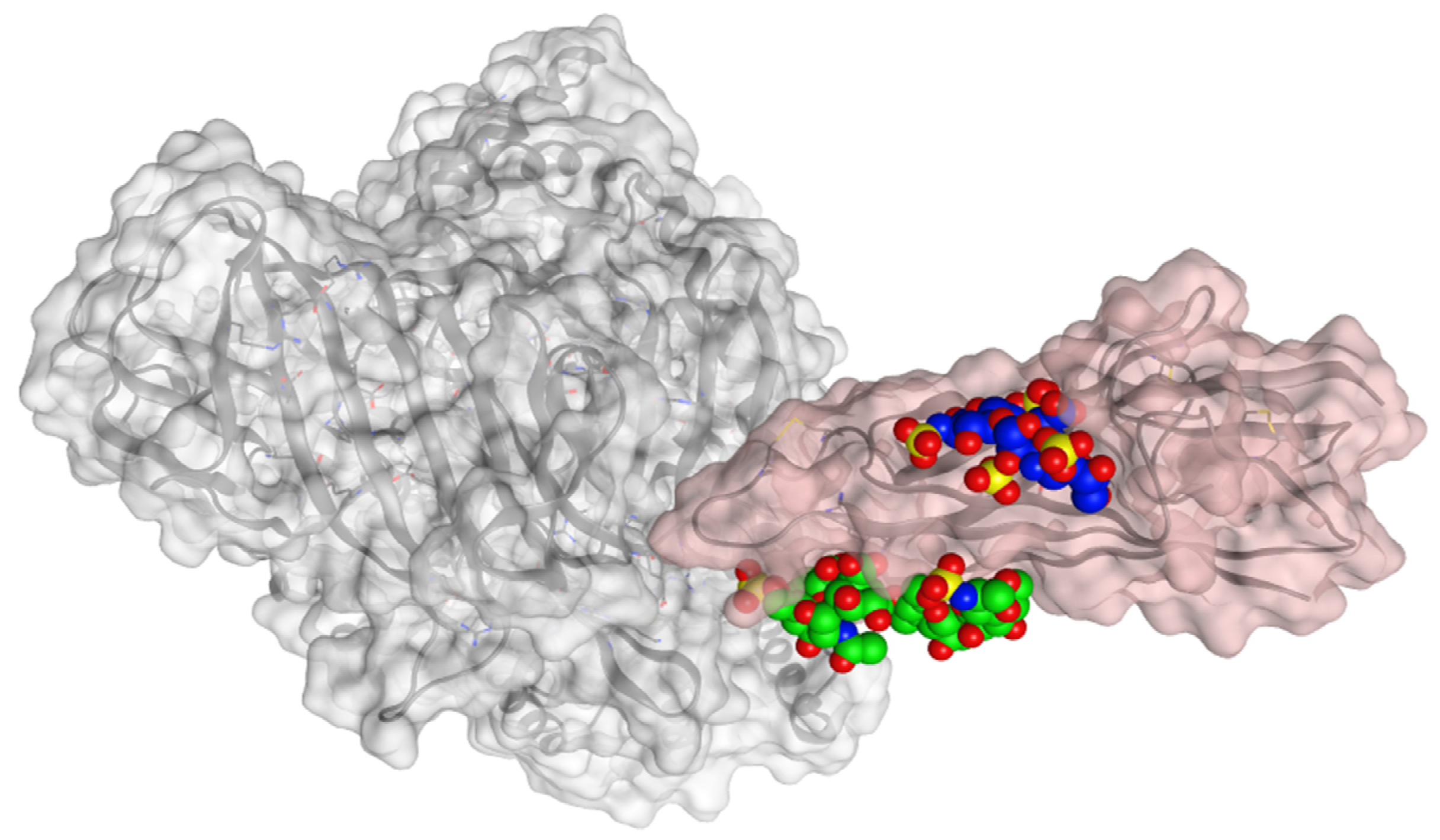
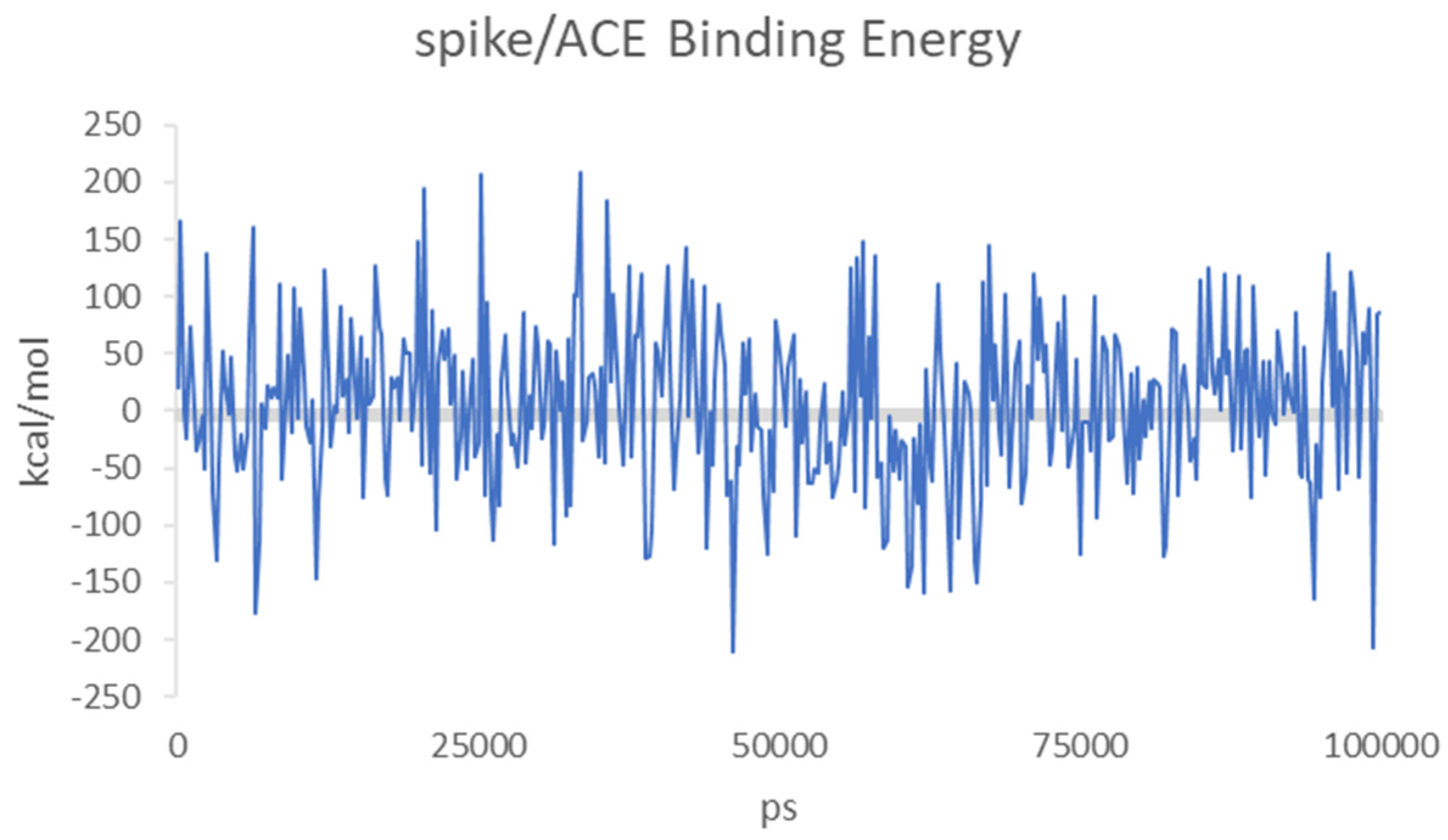
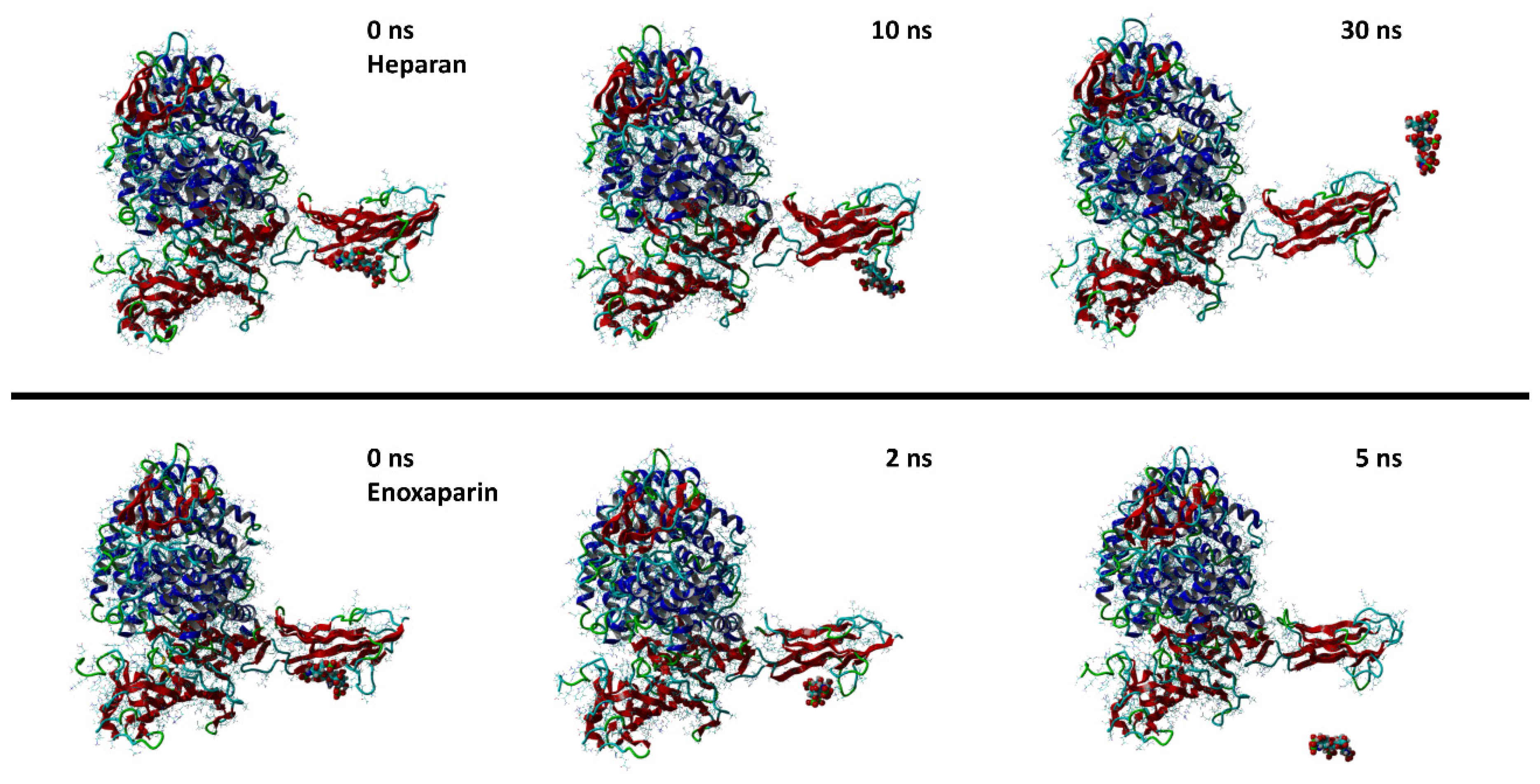
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabricant, J. The early history of infectious bronchitis. Avian Dis. 1998, 42, 648–650. [Google Scholar] [CrossRef]
- Schalk, A.F.; Hawn, M.C. An Apparently New Respiratory Disease of Baby Chicks. J. Amer. Vet. Med. Ass 1931, 78, 413–423. [Google Scholar]
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of a novel type of common-cold virus in organ cultures. Br. Med. J. 1965, 1, 1467–1470. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, K.; Becker, W.B.; Chanock, R.M. Growth in suckling-mouse brain of "IBV-like" viruses from patients with upper respiratory tract disease. Proc. Natl. Acad. Sci. USA 1967, 58, 2268–2273. [Google Scholar] [CrossRef] [Green Version]
- Hamre, D.; Kindig, D.A.; Mann, J. Growth and intracellular development of a new respiratory virus. J. Virol. 1967, 1, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Fattah, S.; Pal, A.; Kaka, N.; Kakodkar, P. History and Recent Advances in Coronavirus Discovery. In In Silico Modeling of Drugs Against Coronaviruses: Computational Tools and Protocols; Roy, K., Ed.; Springer US: New York, NY, USA, 2021; pp. 3–24. [Google Scholar]
- Weiss, S.R. Forty years with coronaviruses. J. Exp. Med. 2020, 217, e20200537. [Google Scholar] [CrossRef] [Green Version]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.M.; Wolthers, K.C.; Wertheim-van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Mizutani, T. A novel coronavirus, MERS-CoV. Uirusu 2013, 63, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes. Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Jaimes, J.A.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2021, 45, fuaa057. [Google Scholar] [CrossRef]
- Nassar, A.; Ibrahim, I.M.; Amin, F.G.; Magdy, M.; Elgharib, A.M.; Azzam, E.B.; Nasser, F.; Yousry, K.; Shamkh, I.M.; Mahdy, S.M.; et al. A Review of Human Coronaviruses’ Receptors: The Host-Cell Targets for the Crown Bearing Viruses. Molecules 2021, 26, 6455. [Google Scholar] [CrossRef]
- Vafaeinezhad, A.; Atashzar, M.R.; Baharlou, R. The Immune Responses against Coronavirus Infections: Friend or Foe? Int. Arch. Allergy Immunol. 2021, 182, 863–876. [Google Scholar] [CrossRef]
- Velusamy, P.; Kiruba, K.; Su, C.H.; Arun, V.; Anbu, P.; Gopinath, S.C.B.; Vaseeharan, B. SARS-CoV-2 spike protein: Site-specific breakpoints for the development of COVID-19 vaccines. J. King Saud University. Sci. 2021, 33, 101648. [Google Scholar] [CrossRef]
- Mazzoni, A.; Vanni, A.; Spinicci, M.; Capone, M.; Lamacchia, G.; Salvati, L.; Coppi, M.; Antonelli, A.; Carnasciali, A.; Farahvachi, P.; et al. SARS-CoV-2 Spike-Specific CD4+ T Cell Response Is Conserved Against Variants of Concern, Including Omicron. Front. Immunol. 2022, 13, 121. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [Green Version]
- King, S.B.; Singh, M. Comparative genomic analysis reveals varying levels of mammalian adaptation to coronavirus infections. PLoS Comput. Biol. 2021, 17, e1009560. [Google Scholar] [CrossRef]
- Li, W.; Hulswit, R.J.G.; Widjaja, I.; Raj, V.S.; McBride, R.; Peng, W.; Widagdo, W.; Tortorici, M.A.; van Dieren, B.; Lang, Y.; et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl. Acad. Sci. USA 2017, 114, E8508–E8517. [Google Scholar] [CrossRef] [Green Version]
- Milewska, A.; Zarebski, M.; Nowak, P.; Stozek, K.; Potempa, J.; Pyrc, K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014, 88, 13221–13230. [Google Scholar] [CrossRef] [Green Version]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nat. Biotechnol. 1992, 357, 420–422. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Cardullo, N.; Catinella, G.; Floresta, G.; Muccilli, V.; Rosselli, S.; Rescifina, A.; Bruno, M.; Tringali, C. Synthesis of Rosmarinic Acid Amides as Antioxidative and Hypoglycemic Agents. J. Nat. Prod. 2019, 82, 573–582. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A. Fourfold Filtered Statistical/Computational Approach for the Identification of Imidazole Compounds as HO-1 Inhibitors from Natural Products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Pistarà, V.; Amata, E.; Dichiara, M.; Damigella, A.; Marrazzo, A.; Prezzavento, O.; Punzo, F.; Rescifina, A. Molecular modeling studies of pseudouridine isoxazolidinyl nucleoside analogues as potential inhibitors of the pseudouridine 5ʹ-monophosphate glycosidase. Chem. Biol. Drug Des. 2018, 91, 519–525. [Google Scholar] [CrossRef]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-Approved Drugs for Treating Iatrogenic Botulism: A Paired 3D-QSAR/Docking Approach(†). ChemMedChem 2020, 15, 256–262. [Google Scholar] [CrossRef]
- Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Mar. Drugs 2019, 17, 624. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Rescifina, A.; Abbate, V. Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules. Int. J. Mol. Sci. 2019, 20, 2311. [Google Scholar] [CrossRef] [Green Version]
- Gentile, D.; Floresta, G.; Patamia, V.; Chiaramonte, R.; Mauro, G.L.; Rescifina, A.; Vecchio, M. An Integrated Pharmacophore/Docking/3D-QSAR Approach to Screening a Large Library of Products in Search of Future Botulinum Neurotoxin A Inhibitors. Int. J. Mol. Sci. 2020, 21, 9470. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of "Small" Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varrica, M.G.; Zagni, C.; Mineo, P.G.; Floresta, G.; Monciino, G.; Pistarà, V.; Abbadessa, A.; Nicosia, A.; Castilho, R.M.; Amata, E.; et al. DNA intercalators based on (1,10-phenanthrolin-2-yl)isoxazolidin-5-yl core with better growth inhibition and selectivity than cisplatin upon head and neck squamous cells carcinoma. Eur. J. Med. Chem. 2018, 143, 583–590. [Google Scholar] [CrossRef]
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. FABP4 inhibitors 3D-QSAR model and isosteric replacement of BMS309403 datasets. Data Brief 2019, 22, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Zagni, C.; Pistara, V.; Oliveira, L.A.; Castilho, R.M.; Romeo, G.; Chiacchio, U.; Rescifina, A. Serendipitous discovery of potent human head and neck squamous cell carcinoma anti-cancer molecules: A fortunate failure of a rational molecular design. Eur. J. Med. Chem. 2017, 141, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Coco, A.; Patamia, V.; Zagni, C.; Floresta, G.; Rescifina, A. Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process. Int. J. Mol. Sci. 2022, 23, 10067. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Zagni, C.; Gentile, D.; Patamia, V.; Rescifina, A. Artificial Intelligence Technologies for COVID-19 De Novo Drug Design. Int. J. Mol. Sci. 2022, 23, 3261. [Google Scholar] [CrossRef]
- Gentile, D.; Patamia, V.; Fuochi, V.; Furneri, P.M.; Rescifina, A. Natural Substances in the Fight of SARS-CoV-2: A Critical Evaluation Resulting from the Cross-Fertilization of Molecular Modeling Data with the Pharmacological Aspects. Curr. Med. Chem. 2021, 28, 8333–8383. [Google Scholar] [CrossRef]
- Fuochi, V.; Furneri, P.M. Natural Substances and Semisynthetic Derivatives as Potential Alternative Products Against SARS-CoV-2. Mini Rev. Med. Chem. 2021, 21, 1596–1611. [Google Scholar] [CrossRef]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef] [Green Version]
- Colpitts, C.C.; Baumert, T.F. Cell Surface Glycans as Viral Entry Factors and Targets for Broadly Acting Antivirals. Recent Adv. Biotechnol. Recent Prog. Glycotherapy 2016, 3, 39–59. [Google Scholar]
- Tzanakakis, G.; Kovalszky, I.; Heldin, P.; Nikitovic, D. Proteoglycans/glycosaminoglycans: From basic research to clinical practice. BioMed Res. Int. 2014, 2014, 295254. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Aquino, R.S.; Park, P.W. Coreceptor functions of cell surface heparan sulfate proteoglycans. Am. J. Physiol. Cell Physiol. 2022, 322, C896–C912. [Google Scholar] [CrossRef] [PubMed]
- Tumova, S.; Woods, A.; Couchman, J.R. Heparan sulfate proteoglycans on the cell surface: Versatile coordinators of cellular functions. Int. J. Biochem. Cell Biol. 2000, 32, 269–288. [Google Scholar] [CrossRef]
- Dudas, J.; Bocsi, J.; Fullar, A.; Baghy, K.; Fule, T.; Kudaibergenova, S.; Kovalszky, I. Heparin and Liver Heparan Sulfate Can Rescue Hepatoma Cells from Topotecan Action. BioMed Res. Int. 2014, 2014, 765794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kultti, A.; Zhao, C.M.; Singha, N.C.; Zimmerman, S.; Osgood, R.J.; Symons, R.; Jiang, P.; Li, X.M.; Thompson, C.B.; Infante, J.R.; et al. Accumulation of Extracellular Hyaluronan by Hyaluronan Synthase 3 Promotes Tumor Growth and Modulates the Pancreatic Cancer Microenvironment. BioMed Res. Int. 2014, 2014, 817613. [Google Scholar] [CrossRef] [Green Version]
- Mundt, F.; Heidari-Hamedani, G.; Nilsonne, G.; Metintas, M.; Hjerpe, A.; Dobra, K. Diagnostic and Prognostic Value of Soluble Syndecan-1 in Pleural Malignancies. BioMed Res. Int. 2014, 2014, 419853. [Google Scholar] [CrossRef] [Green Version]
- Siebert, J.R.; Conta Steencken, A.; Osterhout, D.J. Chondroitin sulfate proteoglycans in the nervous system: Inhibitors to repair. BioMed Res. Int. 2014, 2014, 845323. [Google Scholar] [CrossRef] [Green Version]
- Tolg, C.; McCarthy, J.B.; Yazdani, A.; Turley, E.A. Hyaluronan and RHAMM in Wound Repair and the "Cancerization" of Stromal Tissues. BioMed Res. Int. 2014, 2014, 103923. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.L.; Zhang, X.; Wang, X.M.; Li, J.P. Towards Understanding the Roles of Heparan Sulfate Proteoglycans in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 516028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, M.; Ranasinghe, C.; Jackson, R.; Parish, C.R. Heparan sulfate as a receptor for poxvirus infections and as a target for antiviral agents. J. Gen. Virol. 2017, 98, 2556–2568. [Google Scholar] [CrossRef] [PubMed]
- Shriver, Z.; Capila, I.; Venkataraman, G.; Sasisekharan, R. Heparin and heparan sulfate: Analyzing structure and microheterogeneity. In Heparin—A Century of Progress; Springer: Berlin/Heidelberg, Germany, 2012; pp. 159–176. [Google Scholar]
- Liu, L.; Chopra, P.; Li, X.; Bouwman, K.M.; Tompkins, S.M.; Wolfert, M.A.; de Vries, R.P.; Boons, G.J. Heparan Sulfate Proteoglycans as Attachment Factor for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e1015. [Google Scholar] [CrossRef]
- Jupalli, A.; Iqbal, A.M. Enoxaparin; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Solari, F.; Varacallo, M. Low Molecular Weight Heparin (LMWH); StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs. Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients With COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 1612–1620. [Google Scholar] [CrossRef]
- Joannidis, M.; Kountchev, J.; Rauchenzauner, M.; Schusterschitz, N.; Ulmer, H.; Mayr, A.; Bellmann, R. Enoxaparin vs. unfractionated heparin for anticoagulation during continuous veno-venous hemofiltration: A randomized controlled crossover study. Intensive Care Med. 2007, 33, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, M.; Hartig, F.; Pechlaner, C. Enoxaparin vs. unfractionated heparin in acute coronary syndrome. JAMA 2004, 292, 45–54. [Google Scholar]
- Fuochi, V.; Caruso, M.; Emma, R.; Stivala, A.; Polosa, R.; Distefano, A.; Furneri, P.M. Investigation on the antibacterial activity of electronic cigarette liquids (ECLs): A proof of concept study. Curr. Pharm. Biotechnol. 2020, 22, 983–994. [Google Scholar] [CrossRef]
- Caruso, M.; Distefano, A.; Emma, R.; Zuccarello, P.; Copat, C.; Ferrante, M.; Carota, G.; Pulvirenti, R.; Polosa, R.; Missale, G.A.; et al. In vitro cytoxicity profile of e-cigarette liquid samples on primary human bronchial epithelial cells. Drug. Test Anal. 2022. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Patel, K.; Jaunky, T.; Santopietro, S.; Camacho, O.M.; McAughey, J.; Gaca, M. Electronic cigarette aerosol induces significantly less cytotoxicity than tobacco smoke. Toxicol. Mech. Methods 2016, 26, 477–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, D.; Kilford, J.; Payne, R.; Adamson, J.; Scott, K.; Dalrymple, A.; Meredith, C.; Dillon, D. Characterisation of a Vitrocell(R) VC 10 in vitro smoke exposure system using dose tools and biological analysis. Chem. Cent. J. 2013, 7, 146. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Best, S.A.; Merz, K.M., Jr.; Reynolds, C.H. GB/SA water model for the Merck molecular force field (MMFF). J. Mol. Graph Model. 2000, 18, 273–282. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–105. [Google Scholar] [CrossRef] [PubMed]
- Wallbrecher, R.; Verdurmen, W.P.R.; Schmidt, S.; Bovee-Geurts, P.H.; Broecker, F.; Reinhardt, A.; van Kuppevelt, T.H.; Seeberger, P.H.; Brock, R. The stoichiometry of peptide-heparan sulfate binding as a determinant of uptake efficiency of cell-penetrating peptides. Cell. Mol. Life Sci. 2014, 71, 2717–2729. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Li, Z.; Tomlinson, A.C.; Wong, A.H.; Zhou, D.; Desforges, M.; Talbot, P.J.; Benlekbir, S.; Rubinstein, J.L.; Rini, J.M. The human coronavirus HCoV-229E S-protein structure and receptor binding. eLife 2019, 8, e51230. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. In Computational Drug Discovery and Design; Springer: Berlin/Heidelberg, Germany, 2012; pp. 405–421. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Galimberti, M.; Barbera, V.; Guerra, S.; Bernardi, A. Facile functionalization of sp2 carbon allotropes with a biobased Janus molecule. Rubber Chem. Technol. 2017, 90, 285–307. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Wang, X.; Bie, L.; Gao, J. Structural Insights into the Cofactor Role of Heparin/Heparan Sulfate in Binding between the SARS-CoV-2 Spike Protein and Host Angiotensin-Converting Enzyme II. J. Chem. Inf. Model. 2022, 62, 656–667. [Google Scholar] [CrossRef]
- Li, Y.Q.; Liu, D.Q.; Wang, Y.T.; Su, W.Q.; Liu, G.; Dong, W.J. The Importance of Glycans of Viral and Host Proteins in Enveloped Virus Infection. Front. Immunol. 2021, 12, 638573. [Google Scholar] [CrossRef]
- Miller, N.L.; Clark, T.; Raman, R.; Sasisekharan, R. Glycans in Virus-Host Interactions: A Structural Perspective. Front. Mol. Biosci. 2021, 8, 666756. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Doms, R.W. The entry of entry inhibitors: A fusion of science and medicine. Proc. Natl. Acad. Sci. USA 2003, 100, 10598–10602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melby, T.; Westby, M. Inhibitors of viral entry. In Antiviral Strategies; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 177–202. [Google Scholar]
- Agosto, L.M.; Zhong, P.; Munro, J.; Mothes, W. Highly Active Antiretroviral Therapies Are Effective against HIV-1 Cell-to-Cell Transmission. PLoS Pathog. 2014, 10, e1003982. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Marsh, M. Targeting viral entry as a strategy for broad-spectrum antivirals. F1000Res 2019, 8, 1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiardi, G.; Richter, S.; Oreste, P.; Urbinati, C.; Rusnati, M.; Wade, R.C. The binding of heparin to spike glycoprotein inhibits SARS-CoV-2 infection by three mechanisms. J. Biol. Chem. 2022, 298, 101507. [Google Scholar] [CrossRef]
- Caruso, M.; Distefano, A.; Emma, R.; Di Rosa, M.; Carota, G.; Rust, S.; Polosa, R.; Zuccarello, P.; Ferrante, M.; Raciti, G.; et al. Role of Cigarette Smoke on Angiotensin-Converting Enzyme-2 Protein Membrane Expression in Bronchial Epithelial Cells Using an Air-Liquid Interface. Model. Front. Pharmacol. 2021, 12, 652102. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuochi, V.; Floresta, G.; Emma, R.; Patamia, V.; Caruso, M.; Zagni, C.; Ronchi, F.; Ronchi, C.; Drago, F.; Rescifina, A.; et al. Heparan Sulfate and Enoxaparin Interact at the Interface of the Spike Protein of HCoV-229E but Not with HCoV-OC43. Viruses 2023, 15, 663. https://doi.org/10.3390/v15030663
Fuochi V, Floresta G, Emma R, Patamia V, Caruso M, Zagni C, Ronchi F, Ronchi C, Drago F, Rescifina A, et al. Heparan Sulfate and Enoxaparin Interact at the Interface of the Spike Protein of HCoV-229E but Not with HCoV-OC43. Viruses. 2023; 15(3):663. https://doi.org/10.3390/v15030663
Chicago/Turabian StyleFuochi, Virginia, Giuseppe Floresta, Rosalia Emma, Vincenzo Patamia, Massimo Caruso, Chiara Zagni, Federica Ronchi, Celestino Ronchi, Filippo Drago, Antonio Rescifina, and et al. 2023. "Heparan Sulfate and Enoxaparin Interact at the Interface of the Spike Protein of HCoV-229E but Not with HCoV-OC43" Viruses 15, no. 3: 663. https://doi.org/10.3390/v15030663