Abstract
HoBi-like pestivirus (HoBiPeV), classified under Pestivirus H species, is an emerging cattle pathogen of high economic impact. However, the origin and evolution of HoBiPeV are not very clear due to a lack of full genomic sequences from diverse clades. This study aimed to determine full-genome sequences of HoBiPeV strains of three novel clades (c, d and e) and perform full-genome-based genetic and evolutionary analyses. Bayesian phylogenetic analyses herein confirmed the existence and independent evolution of four main HoBiPeV clades (a, c, d and e) globally, with genetic divergence ranging from 13.0% to 18.2%. Our Bayesian molecular clock estimates revealed that HoBiPeV most likely originated in India, with a dated tMRCA of 1938 (1762–2000), evidencing a more recent origin of HoBiPeV. The evolution rate of HoBiPeV was estimated to be 2.133 × 10−3 subs/site/year at full-genome level but varied widely among individual genes. Selection pressure analyses identified most of the positively selected sites in E2. Additionally, 21.8% of the ORF codon sites were found under strong episodic diversifying selection, providing first evidence of negative selection in HoBiPeV evolution. No recombination event was evident for HoBiPeV-c, d and e strains. These findings provide new insights into HoBiPeV origin and evolutionary history for better understanding the epidemiology and host–pathogen interactions and stimulate vaccine research.
1. Introduction
Infection of cattle with bovine pestiviruses causes respiratory, reproductive and enteric diseases and inflicts significant economic losses in the cattle industry around the world. Belonging to the genus Pestivirus under family Flaviviridae, these viruses are classified into three pestivirus species, namely Pestivirus A (bovine viral diarrhoea virus 1, BVDV-1), Pestivirus B (bovine viral diarrhoea virus 2, BVDV-2) and Pestivirus H (HoBi-like pestivirus, HoBiPeV) [1]. Although less widespread globally than BVDV-1 and BVDV-2, HoBiPeV is currently prevalent in many countries. HoBiPeV genomic RNA is approximately 12.3 kb in length and contains a single large open reading frame (ORF) flanked by 5′- and 3′-untranslated regions (UTR). The ORF encodes a polyprotein of approximately 3900 amino acids that is processed into 12–13 proteins (Npro, C (capsid), envelope proteins Erns, E1 and E2 and non-structural proteins p7, NS2, NS3, (NS2–3), NS4A, NS4B, NS5A and NS5B) by viral and cellular proteases [2].
HoBiPeV was discovered in 2004 in Germany as a cell culture contaminant originating from Brazilian foetal bovine serum (FBS) and the strain was named HoBi_D32/00 [3]. Subsequently, HoBiPeV was detected in commercial FBS lots from Brazil, Argentina, Mexico, Canada and Australia [4,5]. Nevertheless, there is evidence that HoBiPeV was present in Brazilian buffalo much earlier in 1996 [6]. Thus far, natural infection of cattle with HoBiPeV has been detected in three continents: South America (Brazil and Argentina), Europe (Italy and Turkey) and Asia (Thailand, India, Bangladesh and China) [5,7,8,9,10,11,12,13,14]. Although HoBiPeV infection displays similar clinical signs to BVDV-1 and BVDV-2 infections, it has been associated with reproductive disease [15,16], severe respiratory disease [9,17], gastroenteritis and economic losses in cattle farms [14,18] and mucosal disease in cattle [19,20,21,22]. Recent reports of HoBiPeV-associated cattle deaths in China [14] and India [22] are a great concern for the global cattle industry as, currently, there is no commercially available vaccine against HoBiPeV. Aside from cattle, natural HoBiPeV infection has also been detected in buffalo [6,7], sheep and goats [23].
Originally, it was thought that HoBiPeVs were genetically less diverse. However, based on 5′-UTR or Npro genomic sequence analysis, HoBiPeV strains have been classified into five distinct genetic lineages or clades (HoBiPeV-a to HoBiPeV-e) thus far [11,22,23,24,25,26]. The strains of the HoBiPeV-a clade have been widely detected in infected cattle in Brazil, Italy, Turkey, Thailand and China [4,8,12,13,27], while the strains of HoBiPeV-b clade originated in Bangladesh [10]. Surprisingly, the highly divergent HoBiPeV strains of the other three clades (c, d and e) have been detected in cattle exclusively in India [11,22].
Phylogenetic analysis of pestiviruses is vital for classification of novel and emerging viruses, tracing the origin of outbreaks and revealing their evolutionary history. However, little is known about the evolution of HoBiPeVs, and the evolutionary relationships among HoBiPeVs have not been unambiguously determined due to the lack of adequate full-genome sequence data from different clades with diverse geographical origin. Thus far, full genomic sequences are available only for the strains of HoBiPeV-a clade. Hence, the aim of this study was to determine and analyse the complete-genome sequences of the strains of HoBiPeV-c, HoBiPeV-d and HoBiPeV-e clades and decipher the evolutionary relationships among HoBiPeVs based on all available complete-genome sequences. We performed Bayesian evolutionary analysis, selection pressure analysis and recombination analysis to unravel the origin and evolution of HoBiPeVs. Based on Bayesian analysis of the time of most recent common ancestor (TMRCA), we show that HoBiPeV most likely originated in India, with the emergence dating to 1938 (1762–2000). Additionally, we show that HoBiPeVs most likely evolved independently into four clades, and >10.0% divergence at the full-genome level may be considered for classification of genetic clades of HoBiPeVs. Furthermore, we determined the effect of purifying selection, which was uniformly spread over all the genomic regions, in shaping HoBiPeV evolution. The findings here expand our knowledge about the origin and evolutionary history of HoBiPeV.
2. Materials and Methods
2.1. Virus Strains
The complete-genome sequences of the three novel HoBiPeV strains (IndBHA5309/12, IndABI15385/12 and Ind/TN-1214/19) originating from cattle in India were determined and analysed in this study. HoBiPeV IndBHA5309/12 is an archived strain isolated from clinical samples collected from cattle in western India in 2012 and grown in Madin–Darby bovine kidney (MDBK) cells [11]. HoBiPeV IndABI15385/12 strain was isolated from clinical samples from cattle in central India in 2012 and cultured in MDBK cells [11]. HoBiPeV Ind/TN-1214/19 strain was isolated from crossbred Jersey cattle showing mucosal-disease-like lesions in southern India in 2019 and cultured in MDBK cells [22]. These strains represented all three highly divergent HoBiPeV clades (HoBiPeV-c, d and e) reported from India [11,22].
2.2. Next-Generation Sequencing, De Novo Assembly and Genome Annotation
The RNA obtained from low-passage (3rd passage) HoBiPeV strains in MDBK cells were sequenced using an Illumina NextSeq 500 platform (Illumina, San Diego, CA, USA) through outsourcing (M/s Genotypic Technology Pvt. Ltd., Bengaluru, India and Eurofins genomics India, Bengaluru). RNA extraction was performed using the TruSeq RNA Sample Preparation Kit (Illumina). Library preparation was performed following the IlluminaTruSeq RNA library protocol outlined in “TruSeq RNA Sample Preparation Guide” (Illumina). Following second-strand cDNA synthesis, the cDNA was cleaned up using HighPrep PCR (MAGBIO) and Illumina adapters were ligated to the cDNA molecules after end repair and adenylation of the cDNA ends. The prepared library was quantified using Qubit and validated for quality by running an aliquot on High Sensitivity Bioanalyzer Chip (Agilent, Santa Clara, CA, USA). The Illumina paired end (2 × 150 bp) raw reads were quality-checked using FastQC [28]. Illumina raw reads were processed by an in-house script (SeqQC) for adapters and low-quality bases trimming towards 3’-end.
Illumina reads were assembled using SPAdes program [29], intended for de novo assembly after error-correction of sequenced reads. Contig generation was completed using Velvet [30], and virus-related contigs were separated from the assembly. Mapping of high-quality reads on reference genomes (FJ040215: Th_Khonkaen/2004, AB871953: D32_00/HoBi) was completed using Bowtie [31]. De novo assembled sequences were also analysed by comparison to reference HoBiPeV sequences from GenBank via the BLASTn and BLASTx tools of the National Center for Biotechnology Information (NCBI) website. Proteins were predicted from the assembled genome using Prodigal [32].
For confirmation of 5′ and 3′-UTR end sequences of HoBiPeV strains generated through Illumina ngs, we additionally employed RNA ligation method prior to RT-PCR, cloning and Sanger sequencing, as per the previously reported method [33]. Nucleotide sequencing (Sanger sequencing) of three independent clones of each DNA fragment was carried out in an automatic DNA sequencer (ABI 3130, Applied Biosystems, MA, USA) at ICAR-NIHSAD, Bhopal. The three complete-genome sequences of HoBiPeVs generated in this study have been deposited in GenBank under accession numbers OQ411019, OQ411020 and OQ411021.
2.3. Maximum Likelihood Analysis
The nucleotide sequences of complete genomes of 20 HoBi-like pestiviruses and 14 reference pestiviruses available from the GenBank database (Supplementary Table S1) and the three complete-genome sequences of HoBiPeVs generated in this study were aligned using the MUSCLE algorithm. The mean and pairwise divergence of the complete genome and various coding regions were then computed. To assess the evolutionary relationships among HoBi-like and other pestiviruses, phylogenetic trees were inferred by maximum likelihood (ML) method based on the nucleotide alignment of the full-length sequences using MEGA software v. 11 [34]. The ML phylogeny was produced under GTR evolution model, with rate variation following a gamma distribution, as determined by model finder, and robustness of tree topology was assessed by bootstrap analysis with 1000 iterations. The nearest-neighbour interchange (NNI) technique was used to conduct a heuristic tree search, and a neighbour-joining (NJ) tree was selected as the starting tree.
2.4. Recombination Analysis
The full-genome sequence alignment of HoBi-like pestiviruses was examined for any signs of recombination using RDP5 software v. 5.30 [35]. The analysis was carried out using various methods available and their default parameters. Recombination events were only deemed proven if they were identified by at least four of the seven available algorithms (RDP, Geneconv, BootScan, MaxChi, Chimaera, SiScan and 3Seq) with default values. The pairwise homoplasy test (PHI test) was additionally utilised to look for recombination event evidence [36]. This test calculates a p-value and evaluates the significance of evolutionary disagreement across locations in an alignment. A p-value of less than 0.01 indicates the presence of recombination evidence. With a low false-positive rate, the PHI test is particularly effective at determining whether recombination has occurred or not across a broad range of sequence divergence.
2.5. Selection Pressure Analysis
Three likelihood approaches were employed to determine positive selection pressure at certain codon sites: the single-likelihood ancestor counting (SLAC) method, the fixed effects likelihood (FEL) method and a Bayesian strategy called FUBAR. The ratio of non-synonymous (dN) to synonymous (dS) substitutions per site (ratio: dN/dS) was used to calculate the strength of selection pressure. In general, posterior probability >0.9 for FUBAR and p < 0.1 for SLAC strongly imply positive selection. The mixed effects model of evolution (MEME) was used to identify the codon sites that were the subject of episodic diversifying selection. Strong evidence of selection was accepted at significance levels (p < 0.05). All the analyses were carried out using the online Datamonkey webserver [37].
2.6. Bayesian Analysis
The reconstructed ML nucleotide trees were utilised in TempEst [38] to produce linear regression plots of the years of sampling versus root-to-tip distance in order to examine the temporal signal. Using time-stamped sequence data with a relaxed and uncorrelated lognormal clock under the Bayesian Markov chain Monte Carlo (MCMC) method, BEAST version 1.10.4 [39] was used to estimate the rates of evolutionary change (nucleotide substitutions per site per year) and times of circulation of the MRCA (years). The GTR + G + I nucleotide substitution model and an exponential coalescent population with default priors were used. The exponential prior offered a limited HPD range and proper parameter convergence among the many coalescent models examined. For ORF and whole genomes, three independent runs of 200 million generations were carried out, their convergence was evaluated and the log and tree files were then combined with the aid of LogCombiner. After a single run of 200 million generations, convergence was observed for specific coding regions. An asymmetrical-state transition model was used to predict discrete-state ancestral reconstruction of viral sampling locations and migration rates between geographic regions [40]. Using TreeAnnotator 1.7.3, the MCMC chains were condensed to reconstruct the MCC trees. FigTree application v.1.4.0 was used to visualise and colour trees. The statistical uncertainty in the parameter estimates across the sampled trees was reflected in the 95% highest probability density (HPD) intervals.
3. Results
3.1. HoBiPeV Genome Characteristics
The complete-genome sequences of three HoBiPeV strains from India were determined in this study and the genomes displayed a classic pestivirus genome organization, comprising one open reading frame (ORF) flanked by untranslated regions (UTRs) at either end, the 5′-UTR and 3′-UTR (Table 1). The lengths of the three complete genomes of Indian HoBiPeVs were determined to be 12,372, 12,251 and 12,259 nt, respectively, for strains of HoBiPeV-c (IndABI15385/2012), HoBiPeV-d (IndBHA5309/2012) and HoBiPeV-e (HoBiPeV Ind/TN-1214/19). The ORF length of all three HoBiPeVs was alike and comprised 11,700 nt, which encoded a polyprotein encompassing 3899 aa. No viral gene duplications or cellular sequence insertions were found in the genomic RNA of these three HoBiPeVs.

Table 1.
Summary of sequencing data of three complete HoBiPeV genomes (clade c, d and e) determined in this study and their comparison with globally circulating HoBiPeV strains in length of genome, polyprotein and individual genes.
The 5′-UTR of IndABI15385/12, IndBHA5309/12 and HoBiPeV Ind/TN-1214/19 consisted of 393 nt, 373 nt and 367 nt, respectively (Table 1). It is noteworthy that the 393 nt length of 5′-UTR of IndABI15385/12 is the highest among the HoBiPeV strains reported so far, which is 2 nt longer than Chinese strain HN1507. The 3′-UTR of IndABI15385/12 consisted of 279 nucleotides with AT content of 68.1%, while 3′-UTR of IndBHA5309/12 consisted of 178 nucleotides with AT content of 64.61% and Ind/TN-1214/19 consisted of 189 nucleotides with AT content of 63.5%. As the length of 3′-UTR has been reported to be variable between 134 and 255 nts for HoBiPeV strains, the 279 nt length of 3′-UTR in IndABI15385/12 found here is the longest among the HoBiPeV strains and other bovine pestiviruses (BVDV-1 and BVDV-2) reported so far and is 15 nt longer than HoBiPeV strain Th/04_KhonKaen (Table 1).
The complete-genome nucleotide sequence identity between IndABI15385/12 (HoBiPeV-c) and IndBHA5309/12 (HoBiPeV-d) was found to be 79.8%, whereas it was 85.1% between IndABI15385/12 (HoBiPeV-c) and Ind/TN-1214/19 (HoBiPeV-e) and 82.3% between IndBHA5309/12 (HoBiPeV-d) and Ind/TN-1214/19 (HoBiPeV-e) (Supplementary Table S2). The amino acid identity at the complete ORF level was 84.5% between IndABI15385/12 (HoBiPeV-c) and IndBHA5309/12 (HoBiPeV-d), whereas it was 89.9% between IndBHA5309/12 (HoBiPeV-d) and Ind/TN-1214/19 (HoBiPeV-e) and 91.3% between IndABI15385/12 (HoBiPeV-c) and Ind/TN-1214/19 (HoBiPeV-e). The nucleotide and amino acid sequence identity between these strains and strains of the HoBiPeV-a clade are shown in Supplementary Table S2. The amino acid mutations unique to the strains of HoBiPeV-c, d and e clades analysed here are shown in Table 2.
Table 2.
Unique amino acid changes observed in Indian HoBiPeV strains of clades c, d and e compared to the HoBiPeV-a strains analysed in this study.
3.2. Phylogenetic Relationships
The complete-genome sequences of three HoBiPeV strains collected from India and determined in this study were aligned with 20 full-genome sequences of HoBi-like viruses and 14 representative strains from other pestivirus species available in GenBank. The analysed HoBiPeV strains were collected between 2000 and 2019 (Table 1). As per the history available, the strains are from six countries or regions, including Brazil (n = 10), China (n = 2), Thailand (n = 1), Italy (n = 6), India (n = 3) and South America (n = 1).
The maximum likelihood (ML) phylogenetic tree (Figure 1) based on whole-genome sequence analyses showed that globally circulating HoBiPeVs can be resolved clearly into four clades (a, c, d and e) and supported by strong bootstrap values (100%). Since the entire genome of the HoBiPeV-b clade is not currently available, it was excluded from the analysis. In the ML tree, the strain of the HoBiPeV-d clade formed an ancestral node and showed percent nt divergence of 17.9–18.7, 18.2–17.7 and 18.2–17.7, respectively, from clades HoBiPeV-a, HoBiPeV-c and HoBiPeV-d, which is the highest inter-clade diversity observed. Strains HoBiPeV-e and HoBiPeV-c showed 12.7–13.4% and 14.3–15.0% divergence, respectively, from strains of clade HoBiPeV-a. Strains HoBiPeV-e and HoBiPeV-c had a nt divergence of 14.8%. The average genetic divergence among HoBiPeV-a clade was found to be 4.0%. Minimum inter-clade mean genetic distance was observed between HoBiPeV-a and HoBiPeV-e (13.0%), and maximum mean genetic distance was observed between HoBiPeV-a and HoBiPeV-d (18.2%). Based on our results, a cut-off of >10.0% divergence at the full-genome level may be considered for classification of genetic clades of HoBi-like pestivirus. For the HoBiPeV genome dataset, percent nt and aa divergence at various coding regions were determined and shown in Table 3. As anticipated, the E2 region had the highest mean nt and aa diversity, followed by the NS5A coding region. With the exception of p7, there was a high degree of phylogenetic congruence between the whole-genome-based ML tree and those built using various gene sections.
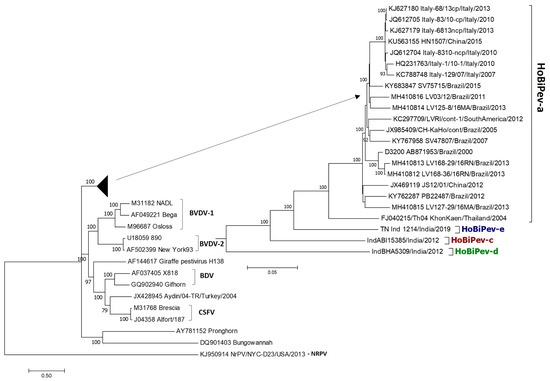
Figure 1.
Phylogenetic tree based on full-genome sequences of HoBiPeV genetic lineages a, c, d and e depicting relationships among HoBiPeVs and between the HoBiPeV and other pestiviruses. Tree topology was assessed by bootstrap analyses with 1000 replicates, and values are indicated in nodes. The sequences of HoBiPeV clades c, d and e were determined in this study and other sequences are from GenBank (Table 1). The scale bar indicates 5% of nucleotide divergence.
Table 3.
Percent nucleotide and amino acid divergence of different coding regions of HoBi-like pestiviruses, including the strains of clades c, d and e analysed in this study.
3.3. Selection Pressure
We simultaneously used three techniques (SLAC, FEL and FUBAR) to analyse the selection pressure acting on the codon sites of the ORF of HoBi-like pestivirus. In general, the calculated dN/dS ratios for the ORF (0.126) and several genes (range from 0.060 to 0.230) of HoBiPeVs were all less than 1.0, indicating modest selection pressure (Table 4). Among the different genomic regions, a higher dN/dS ratio was observed for E2 and the lowest for C. Four codon positions (Npro-114, E2-20, NS5A-304 and NS5B-203) were found to be under positive selection by both SLAC and FUBAR. In addition, positions E2-89 and NS5B-179 were identified by FUBAR and SLAC, respectively. FEL identified 19 sites to be under selection pressure, most of which (11 out of 19) are located within the E2 protein.
Table 4.
HoBiPeV codon sites inferred to be under positive, episodic and negative selection pressure in this study, including the new sequences from HoBiPeV clades c, d and e. Positive selection pressure of codon sites was estimated by the SLAC (single-likelihood ancestor counting), FEL (fixed effects likelihood) and FUBAR (fast unbiased Bayesian approximation) methods. MEME (mixed effects model of evolution) was used to identify the codon sites under episodic diversifying selection. Criteria for strong evidence of selection: significance levels (p < 0.05).
Additionally, the MEME likelihood approach was used to identify sites under episodic diversifying selection. The approach projected episodic pressure at 68 codon sites in HobiPeVs. E2 (n = 15) had the second-highest number of codon sites subject to episodic selection, followed by NS2/3 (n = 20), and 21.8% of codon sites in the ORF were found to be under strong purifying selection, which indicated a major role for negative selection in shaping their evolution. Negatively selected sites were identified using the SLAC method. Further, negative Tajima’s D values (−1.181018) and low nucleotide diversity (0.070774) among the whole-genome sequences of HobiPeV strains point to low frequency of nucleotide polymorphism and strong selection and/or population size expansion. With the exception of Erns, which had the lowest value, our analysis demonstrated that the effect of negative selection was nearly uniformly spread over various genomic regions.
3.4. Evidence of Recombination
We investigated possible recombination events within Indian HoBiPeV genomes and other reference genomes using RDP5 software. The results showed that five recombinant events resulted in emergence of four recombinant viruses, which were detected in the current study with a p-value of 0.01 and a recombinant score of >0.4 (Table 5). HoBiPeV strain Italy-1/10 (HQ231763) identified in Italy was found to be a double recombinant. In most of the cases, it is also possible that the major parent could be the actual recombinant. The recombination sites were distributed non-randomly along the genome and the recombination breakpoints were detected in 5′-UTR, E2, NS2/3, NS4B, NS5A and 3′-UTR. All the recombination events were found only among the strains of HoBiPeV-a clade, and none of the Indian HoBiPeVs (HoBiPeV clade-c, d and e) sequenced in this study showed recombination. The PHI test, on the other hand, did not find statistically significant evidence for recombination (p = 0.09). Nearly the same values were obtained when we performed the evolutionary analysis after excluding putative recombinant sequences, suggesting that the recombinant fragment was shorter and had little effect on the estimate.
Table 5.
Putative recombination events in HoBi-like pestiviruses, including the strains of clades c, d and e sequenced in this study using RDP5 software.
3.5. Temporal and Spatial Structure
Root-to-tip regression analysis of the genetic distances of HoBi-like pestiviruses against sampling time, conducted using TempEst software, produced a correlation coefficient range of 0.31 to 0.45 and coefficient of determination (R2) of 0.09 to 0.20 for the whole genome, the ORF and different coding regions (Table 6). The 5′-UTR region showed the maximum temporal signal, followed by E1, C, NS2/3, Npro and the whole genome. Overall, although not very strong, a positive correlation was observed, thus suggesting a significant relationship between genetic divergence and time. A whole-genome-based maximum clade credibility (MCC) tree reconstructed in this study revealed two main clusters: one contains all the strains of HoBiPeV-a clade except strain Th04_KhonKaen, which, in turn, was placed in a separate cluster along with the strains of HoBiPeV-c, d and e (Figure 2). The E2 gene-derived MCC tree, on the other hand, revealed the same topology as the ML tree.
Table 6.
Estimates from Bayesian MCMC (Markov chain Monte Carlo) analysis for HoBi-like pestivirus genomes, including the genomes of clade c, d and e sequenced in this study. The evolutionary rates, tMRCA and RSPP values corresponding to the geographical locations are shown.
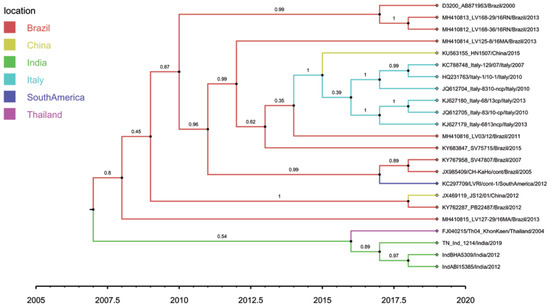
Figure 2.
Tip-labelled maximum clade credibility phylogenetic tree reconstructed from 23 globally distributed HoBiPeV full genomes, including sequences from the three divergent clades (c, d and e) determined in this study. The rates of HoBiPeV evolutionary change (nucleotide substitutions per site per year) and times of circulation of the MRCA (years) were estimated using time-stamped sequence data with a relaxed and uncorrelated lognormal clock under the Bayesian Markov chain Monte Carlo (MCMC) method, BEAST version 1.10.4 [39]. The branches are coloured based on the most probable location of the descendent nodes. The sequences from this study are highlighted with green colour.
The relative nucleotide substitution rates at all three codon positions in ORF showed that substitutions were more frequent at the third codon position (2.242, 95% HPD 2.201–2.286) compared to the first (0.499, 95% HPD 0.461–0.535) and second (0.259, 95% HPD 0.233–0.285), as expected. The mean rate of evolutionary change in the HoBiPeV was estimated to be 2.133 × 10−3 subs/site/year (95% HPDs of 2.181 × 10−4 3.933 × 10−3 subs/site/year) for the full genome and 2.074 × 10−3 subs/site/year (95% HPDs of 2.833 × 10−4 3.759 × 10−3 subs/site/year) for the ORF (Table 6). The substitution rate of each coding region of HoBiPeV was compared and the estimates varied in the following order: E2, Erns, NS5B, NS4A, Npro, NS4B, NS2/3, NS5A, E1, C and P7. Surprisingly, the E2 coding region had a nucleotide substitution rate considerably lower than other coding regions and the whole genome. The P7 protein coding region was found to have the highest evolutionary rate, and, among the structural coding regions, the C region showed the fastest evolutionary rate followed by the E1 region.
The results of our Bayesian phylogenetic analysis based on whole-genome sequences (Table 6) suggested that HoBi-like viruses most likely emerged in India (root state posterior probability (RSPP) = 42.33%), with a recent common ancestor in 1938 CE (95% credibility interval: 1762–2000 CE). When ORF alone was used in the analysis, HoBiPeVs from India received an RSPP of 40.86% and HoBiPeVs found in Brazil received 39.69%, with a tMRCA of 1942 CE (95% credibility interval, 1800–1998 CE). Similar to this, the estimated tMRCA derived from individual gene sequences covered the period from 1962 to 1989, with the exception of the Npro and E2 genes, which yielded estimates from much earlier periods. In our analysis, the tMRCA estimate for the E2 gene dates to the 12th century and that for the Npro region to the 18th century.
4. Discussion
HoBi-like pestiviruses are a serious emerging threat to cattle populations, but only a few full-genome-sequence-based studies are available. Thus far, HoBiPeV strains have been classified into five clades or genotypes (a–e) based on partial 5′-UTR or Npro sequences, but full genomic sequences are available only for strains of the HoBiPeV-a clade. This work was conducted to genetically characterise the representative strains from HoBiPeV clades c, d and e from India at the entire-genome level and determine the evolutionary relationships of HoBiPeVs because strains of three out of the five lineages described thus far have been circulating in India. Here, we report the first complete-genome sequences of strains belonging to the HoBiPeV-c, HoBiPeV-d and HoBiPeV-e clades and the full-genome-based evolutionary analyses of HoBiPeVs.
Use of at least two-genetic-regions-based phylogenies is advised for classification of HoBiPeV lineages, and trees-based datasets combining the 5′-UTR and Npro genetic regions or 5′-UTR-Npro-E2 provide a robust phylogeny with higher statistical support [11,24]. The previously described HoBiPeVs from Brazil, Italy and Thailand were found to be very closely related and have formed a single genetic lineage, known as the HoBiPeV-a clade [24,26]. Subsequently, the HoBiPeV strains from cattle in Bangladesh were assigned to the HoBiPeV-b clade [10]. The strains of two highly divergent and novel linages from India were then described in 2014 and named the HoBiPeV-c and -d clades based on concatenated datasets of the 5′-UTR and Npro sequences [11]. Recently, it has been found that India is home to strains of yet another unique lineage, the HoBiPeV-e clade [22]. However, evolutionary analyses between bovine pestiviruses and among HoBiPeVs have not been deciphered clearly as different phylogenetic relationships are inferred from analysis of different genetic regions [41] and for HoBiPeV. Thus far, only a few studies are available based on complete-genome-based phylogenetic analysis of strains belonging to only the HoBiPeV-a clade [26,42].
In the present study, we showed that the HoBiPeV clade formed a sister-clade with two other bovine pestivirus species, BVDV-1 and BVDV-2, based on phylogenetic analysis of full-length pestivirus genomic sequences, including the complete genomic sequences of four HoBiPeV clades, confirming the earlier hypothesis that these bovine pestiviruses have emerged from a common ancestor [24,41,42]. Furthermore, based on whole-genome sequence analyses, this study confirms the existence of four HoBiPeV main clades (a, c, d and e) globally, with the HoBiPeV-d strain found in India forming an ancestral node. The average genetic divergence among the strains of the HoBiPeV-a clade was found to be 4.0%, which is inconsistent with a previous study [26]. However, our results here showed the minimum inter-clade mean genetic divergence between HoBiPeV-a and HoBiPeV-e (13.0%) and the maximum mean genetic divergence between HoBiPeV-a and HoBiPeV-d (18.2%), suggesting that a cut-off of >10.0% divergence at the full-genome level may be considered for classification of genetic clades of HoBi-like pestiviruses.
The origin and evolutionary divergence of HoBiPeV have been a matter of debate recently. In this study, we performed a Bayesian molecular clock estimate based on full-genome sequences from four HoBiPeV main clades, showing that HoBiPeV most likely originated in India with a tMRCA of 1938 CE (95% credibility interval: 1762–2000 CE) approximately. Additionally, based on ORF analysis, HoBiPeVs revealed a tMRCA of 1942 CE (95% credibility interval, 1800–1998 CE), while the estimated tMRCA derived from individual gene sequences covered the period from 1962 to 1989, with the exception of the Npro (18th century) and E2 (12th century). The studies on evolutionary analyses with regard to the origin and dating of tMRCA of HoBiPeVs have so far been based on either single-gene, 5′-UTR [43], Npro [26] or combined datasets of 5′-UTR-Npro-E2 [24] wherein various estimates of tMRCA were reported. For instance, a previous study [24] reported a tMRCA of 1880 (HPDs, 1651–1993) based on combined datasets of 5′-UTR, Npro and E2 gene. In contrast, another study [26] reported a tMRCA of 1566 (95% HPD 1001–1901) based on the Npro gene, while a recent study [43] reported a tMRCA of 1952 (95% HPD 1905–1985) based on the 5′-UTR.
The dated tMRCAs of earliest origin, 1566 or 1880, suggested in previous studies [24,26], are much earlier than the date (1938) we estimated in this study. Meanwhile, the origin date of 1952 speculated by a recent study [43] is closest to the date of origin estimated in our study. This could be because previous Bayesian analyses used mostly HoBiPeV-a sequences or included some available 5′-UTR/Npro sequences from clades c and d [24,26,43]. In contrast, this study is based on Bayesian analysis of full-genome sequences of the HoBiPeV strains of all four main clades (a, c, d and e). In addition, our analyses show that the genomic regions chosen for estimating age of tMRCA, along with quantity of sequences and time period, have a significant influence on the estimates. It is important to keep in mind that the majority of reported dates of tMRCA, including those reported in this study, are linked to broad HPDs; hence, it is likely that the mean tMRCA date does not accurately represent the actual date. Nevertheless, our findings provide evidence of a more recent origin of HoBiPeV, which has evolved independently into four clades and adds credence to the earlier notion that HoBiPeV most likely originated in India before its emergence in other parts of the world [22,26,43]. A limitation of this study is the sparse evolutionary comparison of sequences at the full-genome level due to their unavailability in public databases, and, hence, more complete-genome-based analyses will produce more reliable and accurate estimates and greater evolutionary knowledge in the future.
Although several hypotheses have been made, the dispersal of HoBiPeV following its origin is not very clear so far. The association of HoBiPeV lineages with geographic regions found earlier and in this study suggests that HoBiPeVs were circulating in cattle populations long before first being discovered [11,26]. Previous studies hypothesized that HoBiPeV evolved independently in the Indian subcontinent and elsewhere [11,25] or was introduced into Brazil coinciding with intensive importation of water buffalo and indicine cattle (Bos indicus) from Asia in the 20th century [26]. A recent study based on Bayesian analysis speculated that Italy is the hub of spread of HoBiPeV as it is the junction between Europe and Asia for export and import [43]. However, due to the range of genetic divergence found at the full-genome level between the HoBiPeV strains (clades c, d and e) circulating in India and in other parts of the world (clade a) in this study and the lack of full-proof epidemiological links, we suppose that, although HoBiPeV most likely originated in India and evolved independently, its spread to different geographical regions remains inconclusive. Indeed, India has a long trade history of cattle germplasm imports from Brazil and several European countries for upgrading the native cattle breeds (Bos indicus). Moreover, despite the high prevalence of HoBiPeV in Brazil, only strains of the HoBiPeV-a clade with little genetic variation have been identified to date [44].
Assessment of the rates of evolution of HoBiPeVs in this study revealed that the mean nucleotide substitution rate of HoBiPeVs is 2.133 × 10−3 subs/site/year (95% HPDs of 2.181 × 10−4 3.933 × 10−3 subs/site/year) for the full genome and 2.074 × 10−3 subs/site/year (95% HPDs of 2.833 × 10−4 3.759 × 10−3 subs/site/year) for the ORF, while the rates varied widely at the individual-gene level. Although the evolution rate for HoBiPeV at the full-genome level is not available so far for comparison, the rate calculated in our study approximates with the evolutionary rate (1.4 × 10−3 subs/site/year) reported for BVDV-1 and BVDV-2 at the full-genome level [45]. In contrast, a relatively higher rate of evolution (approximately 1.1 × 10−2 subs/site/ year) for HoBiPeV has been reported in a previous study [43], which may be because the previous study was based on evolutionary analysis of 5′-UTR sequences. Interestingly, based on individual gene analysis, we found that gene-coding P7 protein has the highest evolutionary rate, and, among the genes of structural coding regions, the C gene showed the fastest evolutionary rate and gene-coding immunodominant E2 protein showed the lowest evolution rate among the coding region genes. The observed low evolutionary rate of E2 is perplexing as it is the most variable gene compared to other coding region genes.
Another focus of our study was selection pressure analysis of HoBiPeV ORF, including the envelope glycoprotein E2 gene, which is immunodominant and generates virus-neutralizing antibodies in the host. We observed modest selection pressure for HoBiPeV as the calculated dN/dS ratios for the ORF (0.126) and several genes (ranging from 0.060 to 0.230) of HoBiPeVs were all <1.0. However, among the different genomic regions, a higher dN/dS ratio was observed for E2, and most of the sites under positive selection pressure (11 of 19) are located within the E2 protein. In a previous study also, HoBiPeV E2 was reported to have the highest number of sites (11 of 18) under positive selection pressure [26]. Our findings also agree with the results of previous studies on two other related bovine pestiviruses, BVDV-1 and BVDV-2 [46,47]. Additionally, we conducted episodic selection pressure analysis of HoBiPeV as, in addition to positive selection, many codon sites may also experience selection in a restricted number of branches, designated as episodic diversifying selection, associated with selection pressure at the host–pathogen interface [48]. Our results showed episodic pressure at 68 codon sites in HoBiPeVs, of which NS2/3 (n = 20) had the highest and E2 (n = 15) had the second-highest number of codon sites subject to episodic selection. Interestingly, 21.8% of the codon sites in the ORF were found to be under strong purifying selection, suggesting a major role for negative selection in shaping HoBiPeV evolution. Episodic diversifying selection has been shown to play a major role in shaping evolution of foot and mouth disease virus (FMDV) VP1 and VP3 [49]. The present study reports a role of episodic diversifying selection in HoBiPeV evolution for the first time. However, identifying the importance of this selection requires further research.
Genetic recombination is an important source of genetic variability of viruses and essential for evolution of most RNA viruses. In this study, based on RDP5 analysis, five potential recombinant events distributed non-randomly along the HoBiPeV genome were found only among the strains of HoBiPeV-a clade, which resulted in emergence of four recombinant viruses. Our findings here are inconsistent with a recent study, where the recombination events were found only among the strains of HoBiPeV-a clade [43]. The only difference was that six potential recombinant events were found in place of five found here as HoBiPeV strain Italy-1/10 (HQ231763) was found to be a triple recombinant in the previous study [43]. This may be due to the differences in analyses methods used in both studies. However, no recombination event was found among the analysed strains of HoBiPeV-c, d and e clades. Although uncommon, both heterologous and homologous recombination events (between different genotypes) have been reported for pestiviruses BVDV-1, BVDV-2 and CSFV under natural replication [50]. Hence, further studies may elucidate whether the same is also true for HoBiPeV.
5. Conclusions
In this study, we determined full genomic sequences of HoBiPeV strains of three novel and divergent clades (c, d and e) circulating in India and conducted the first full-genome-sequence-based genetic and evolutionary analyses of HoBiPeVs. Phylogenetic analyses confirmed the existence and independent evolution of four HoBiPeV main clades (a, c, d and e) globally, with inter-clade genetic divergence rates ranging between 13.0% and 18.2%. Our Bayesian molecular clock estimates revealed that HoBiPeV most likely originated in India, with a dated tMRCA of 1938 (1762–2000), suggesting a more recent origin of HoBiPeV. The rate of evolution of HoBiPeVs was estimated to be 2.133 × 10−3 subs/site/year for the full genome, whereas the evolution rate varied widely at the individual-gene level. Selection pressure analyses showed evidence of strong purifying (negative) selection, suggesting an important role of negative selection in HoBiPeV evolution for the first time. However, we also identified positively selected sites, mostly in E2. The findings of this study provide new insights into HoBiPeV origin and evolutionary history that may benefit in understanding host–pathogen interactions and developing newer therapeutics and vaccines for control of this emerging bovine pestivirus.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/v15030733/s1, Table S1: Full-genome sequences of HoBiPeV strains and other reference pestiviruses used in the dataset for genetic and evolutionary analyses in this study. Table S2: Nucleotide and amino acid sequence identity (%) of Indian HoBiPeV strains of clades c, d and e sequenced in this study with HoBiPeV-a strains and reference pestiviruses.
Author Contributions
Conceptualization, N.M., S.K. and S.S.; Data curation, S.K., D.M., N.M. and S.S.; Methodology, S.K., D.M., N.M., S.S. and S.B.S.; Data analysis and Result interpretation, S.S., S.K., D.M. and N.M.; Supervision, N.M., V.P.S. and A.S.; Writing—original draft, N.M. and S.S.; Review and Editing of Manuscript, N.M., S.S. and S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by a grant (Grant No. IXX08441) from the Institute ICAR-National Institute of High Security Animal Diseases, Bhopal, 462022, India.
Institutional Review Board Statement
Not applicable as no animals were handled in this study.
Informed Consent Statement
Not applicable.
Data Availability Statement
All required data are available as texts and figures in main text of the article or in the Supplementary Materials. The sequence data generated in this study were submitted to GenBank and are available under Accession Numbers OQ411019–OQ411021.
Acknowledgments
The authors thank the Director, Deemed University, ICAR-IVRI, Izatnagar, Bareilly, U.P. for the help as part of this work was carried out by D.M. during his PhD thesis research work at ICAR-NIHSAD, Bhopal. We also thank the Director, ICAR-NIHSAD, Bhopal for administrative and technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smith, D.B.; Meyers, G.; Bukh, J.; Gould, E.A.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; Simmonds, P.; et al. Proposed revision to the taxonomy of the genus Pestivirus, family Flaviviridae. J. Gen. Virol. 2017, 98, 2106–2112. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 6, pp. 712–746. [Google Scholar]
- Schirrmeier, H.; Strebelow, G.; Depner, K.; Hoffmann, B.; Beer, M. Genetic and antigenic characterization of an atypical pestivirus isolate, a putative member of a novel pestivirus species. J. Gen. Virol. 2004, 85, 3647–3652. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Vijayaraghavan, B.; Belak, S.; Liu, L. Detection and identification of the atypical bovine pestiviruses in commercial foetal bovine serum batches. PLoS ONE 2011, 6, e28553. [Google Scholar] [CrossRef] [PubMed]
- Pecora, A.; Perez Aguirreburualde, M.S.; Ridpath, J.F.; Dus Santos, M.J. Molecular characterization of pestiviruses in fetal bovine sera originating from Argentina: Evidence of circulation of HoBi-like viruses. Front. Vet. Sci. 2019, 6, 359. [Google Scholar] [CrossRef]
- Stalder, H.P.; Meier, P.H.; Pfaffen, G.; Wageck-Canal, C.; Rüfenacht, J.; Schaller, P.; Bachofen, C.; Marti, S.; Vogt, H.R.; Peterhans, E. Genetic heterogeneity of pestiviruses of ruminants in Switzerland. Prev. Vet. Med. 2005, 72, 37–41. [Google Scholar] [CrossRef]
- Cortez, A.; Heinemann, M.B.; Castro, A.M.M.G.D.; Soares, R.M.; Pinto, A.M.V.; Alfieri, A.A.; Flores, E.F.; Leite, R.C.; Richtzenhain, L.J. Genetic characterization of Brazilian bovine viral diarrhea virus isolates by partial nucleotide sequencing of the 5′-UTR region. Pesqui. Vet. Bras. 2006, 26, 211–216. [Google Scholar] [CrossRef]
- Ståhl, K.; Kampa, J.; Alenius, S.; Wadman, A.; Baule, C.; Aiumlamai, S.; Belák, S. Natural infection of cattle with an atypical ‘ HoBi ’-like pestivirus—Implications for BVD control and for the safety of biological products. Vet. Res. 2007, 38, 517–523. [Google Scholar] [CrossRef]
- Decaro, N.; Lucente, M.S.; Mari, V.; Cirone, F.; Cordioli, P.; Camero, M.; Buonavoglia, C. Atypical pestivirus and severe respiratory disease in calves, Europe. Emerg. Infect. Dis. 2011, 17, 1549–1552. [Google Scholar] [CrossRef]
- Haider, N.; Rahman, M.S.; Khan, S.U.; Mikolon, A.; Gurley, E.S.; Osmani, M.G.; Shanta, I.S.; Paul, S.K.; Macfarlane-Berry, L.; Islam, A.; et al. Identification and epidemiology of a rare HoBi-like pestivirus strain in Bangladesh. Transbound. Emerg. Dis. 2014, 61, 193–198. [Google Scholar] [CrossRef]
- Mishra, N.; Rajukumar, K.; Pateriya, A.; Kumar, M.; Dubey, P.; Behera, S.P.; Verma, A.; Bhardwaj, P.; Kulkarni, D.D.; Vijaykrishna, D.; et al. Identification and molecular characterization of novel and divergent HoBi-like pestiviruses from naturally infected cattle in India. Vet. Microbiol. 2014, 174, 239–246. [Google Scholar] [CrossRef]
- Timurkan, M.Ö.; Aydın, H. Increased genetic diversity of BVDV strains circulating in Eastern Anatolia, Turkey: First detection of BVDV-3 in Turkey. Trop. Anim. Health. Prod. 2019, 51, 1953–1961. [Google Scholar] [CrossRef]
- Shi, H.; Li, H.; Zhang, Y.; Yang, L.; Hu, Y.; Wang, Z.; Duan, L.; Leng, C.; Yan, B.; Yao, L. Genetic diversity of bovine pestiviruses detected in backyard cattle farms between 2014 and 2019 in Henan Province, China. Front. Vet. Sci. 2020, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, M.; Liu, S.; Shang, Y. HoBi-like pestivirus infection leads to bovine death and severe respiratory disease in China. Transbound. Emerg. Dis. 2021, 68, 1069–1074. [Google Scholar] [CrossRef]
- Decaro, N.; Lucente, M.S.; Mari, V.; Sciarretta, R.; Pinto, P.; Buonavoglia, D.; Martella, V.; Buonavoglia, C. ‘Hobi’-like pestivirus in aborted bovine fetuses. J. Clin. Microbiol. 2012, 50, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N. HoBi-like pestivirus and reproductive disorders. Front. Vet. Sci. 2020, 7, 622447. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Mari, V.; Pinto, P.; Stella Lucente, M.; Sciarretta, R.; Cirone, F.; Colaianni, M.L.; Elia, G.; Thiel, H.J.; Buonavoglia, C. Hobi-like pestivirus: Both biotypes isolated from a diseased animal. J. Gen. Virol. 2012, 93, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Lucente, M.S.; Losurdo, M.; Larocca, V.; Elia, G.; Occhiogrosso, L.; Buonavoglia, C. HoBi-like pestivirus and its impact on cattle productivity. Transbound. Emerg. Dis. 2016, 63, 469–473. [Google Scholar] [CrossRef]
- Decaro, N.; Lanave, G.; Lucente, M.S.; Mari, V.; Varello, K.; Losurdo, M.; Larocca, V.; Bozzetta, E.; Cavaliere, N.; Martella, V.; et al. Mucosal disease-like syndrome in a calf persistently infected by Hobi-like pestivirus. J. Clin. Microbiol. 2014, 52, 2946–2954. [Google Scholar] [CrossRef]
- Weber, M.N.; Mósena, A.C.S.; Simões, S.V.D.; Almeida, L.L.; Pessoa, C.R.M.; Budaszewski, R.F. Clinical presentation resembling mucosal disease associated with ’HoBi’-like pestivirus in a field outbreak. Transbound. Emerg. Dis. 2016, 63, 92–100. [Google Scholar] [CrossRef]
- Cruz, R.A.S.; Rodrigues, W.B.; Silveira, S.; Oliveira, V.H.S.; Campos, C.G.; Leite Filho, R.V.; Boabaid, F.M.; Driemeier, D.; Canal, C.W.; Alfieri, A.A.; et al. Mucosal disease-like lesions caused by HoBi-like pestivirus in Brazilian calves in 2010–2011: Clinical, pathological, immunohistochemical, and virological characterization. Res. Vet. Sci. 2018, 119, 116–121. [Google Scholar] [CrossRef]
- Kalaiyarasu, S.; Mishra, N.; Jayalakshmi, K.; Selvaraj, P.; Sudhakar, S.B.; Jhade, S.K.; Sood, R.; Premalatha, N.; Singh, V.P. Molecular characterization of recent HoBi-like pestivirus isolates from cattle showing mucosal disease-like signs in India reveals emergence of a novel genetic lineage. Transbound. Emerg. Dis. 2022, 69, 308–326. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kan, Y.; Yao, L.; Leng, C.; Tang, Q.; Ji, J.; Sun, S. Identification of Natural Infections in Sheep/Goats with HoBi-like Pestiviruses in China. Transbound. Emerg. Dis. 2016, 63, 480–484. [Google Scholar] [CrossRef]
- Liu, L.; Xia, H.; Wahlberg, N.; Belák, S.; Baule, C. Phylogeny, classification and evolutionary insights into pestiviruses. Virology 2009, 15, 351–357. [Google Scholar] [CrossRef]
- Giammarioli, M.; Ridpath, J.F.; Rossi, E.; Bazzucchi, M.; Casciari, C.; De Mia, G.M. Genetic detection and characterization of emerging HoBi-like viruses in archival foetal bovine serum batches. Biologicals 2015, 43, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Silveira, S.; Cibulski, S.P.; Junqueira, D.M.; Mósena, A.C.S.; Weber, M.N.; Mayer, F.Q.; Canal, C.W. Phylogenetic and evolutionary analysis of HoBi-like pestivirus: Insights into origin and dispersal. Transbound. Emerg. Dis. 2020, 67, 1909–1917. [Google Scholar] [CrossRef]
- Kampa, J.; Alenius, S.; Emanuelson, U.; Chanlun, A.; Aiumlamai, S. Bovine herpesvirus type 1 (BHV-1) and bovine viral diarrhoea virus (BVDV) infections in dairy herds: Self clearance and the detection of seroconversions against a new atypical pestivirus. Vet. J. 2009, 182, 223–230. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 30 August 2021).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Behera, S.P.; Mishra, N.; Vilcek, S.; Rajukumar, K.; Nema, R.K.; Prakash, A.; Kalaiyarasu, S.; Dubey, S.C. Genetic and antigenic characterization of bovine viral diarrhoea virus type 2 isolated from cattle in India. Comp. Immunol. Microbiol. Infect. Dis. 2011, 34, 189–196. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus. Evol. 2020, 7, veaa087. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Liu, L.; Xia, H.; Baule, C.; Belak, S.; Wahlberg, N. Effects of methodology and analysis strategy on robust ness of pestivirus phylogeny. Virus. Res. 2010, 147, 47–52. [Google Scholar] [CrossRef]
- Mósena, A.C.S.; Weber, M.N.; Cibulski, S.P.; Silveira, S.; Silva, M.S.; Mayer, F.Q.; Canal, C.W. Genomic characterization of a bovine viral diarrhea virus subtype 1i in Brazil. Archi. Virol. 2017, 162, 1119–1123. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, L.; Niu, L.; Shangguan, H.; Huang, C.; Yi, Y.; Zhang, Y.; Gao, M.; Ge, J. Genetic and evolutionary analysis of emerging HoBi-like pestivirus. Res. Vet. Sci. 2021, 137, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Bauermann, F.V.; Ridpath, J.F. Epidemiology of Pestivirus H in Brazil and its control implications. Front. Vet. Sci. 2021, 8, 693041. [Google Scholar] [CrossRef] [PubMed]
- Chernick, A.; Meer, F.V. Evolution of bovine viral diarrhea virus in Canada from 1997–2013. Virology 2017, 509, 232–238. [Google Scholar] [CrossRef]
- Lang, Y.; Gao, S.; Du, J.; Shao, J.; Cong, G.; Lin, T.; Zhao, F.; Liu, L.; Chang, H. Polymorphic genetic characterization of E2 gene of bovine viral diarrhea virus in China. Vet. Microbiol. 2014, 174, 554–559. [Google Scholar] [CrossRef]
- Chernick, A.; Ambagala, A.; Orsel, K.; Wasmuth, J.D.; van Marle, G.; van der Meer, F. Bovine viral diarrhea virus genomic variation within persistently infected cattle. Infect. Genet. Evol. 2018, 58, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Subramaniam, S.; Mohapatra, J.K.; Das, B.; Sharma, G.K.; Biswal, J.K.; Mahajan, S.; Misri, J.; Dash, B.B.; Pattnaik, B. Capsid coding region diversity of re-emerging lineage C foot-and-mouth disease virus serotype Asia1 from India. Arch. Virol. 2015, 160, 1751–1759. [Google Scholar] [CrossRef]
- Weber, M.N.; Streck, A.F.; Silveira, S.; M´osena, A.; da Silva, M.S.; Canal, C.W. Homologous recombination in pestiviruses: Identification of three putative novel events between different subtypes/genogroups. Infect. Genet. Evol. 2015, 30, 219–224. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).