
Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling

 , ,
, ,  , , , ,
, , , ,  , , and
, , and
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. SARS-CoV-2 Isolate
2.3. SARS-CoV-2 Challenge
2.4. Viral Quantification
2.5. RNA Libraries and Sequencing (RNA-Seq)
2.6. RNA-Seq Data Analysis
2.7. Downstream RNA-Seq Analysis
2.8. RT-qPCR
2.9. Western Blotting
2.10. Immunofluorescence
2.11. Quantitative Analysis of Mitochondrial Network Morphology
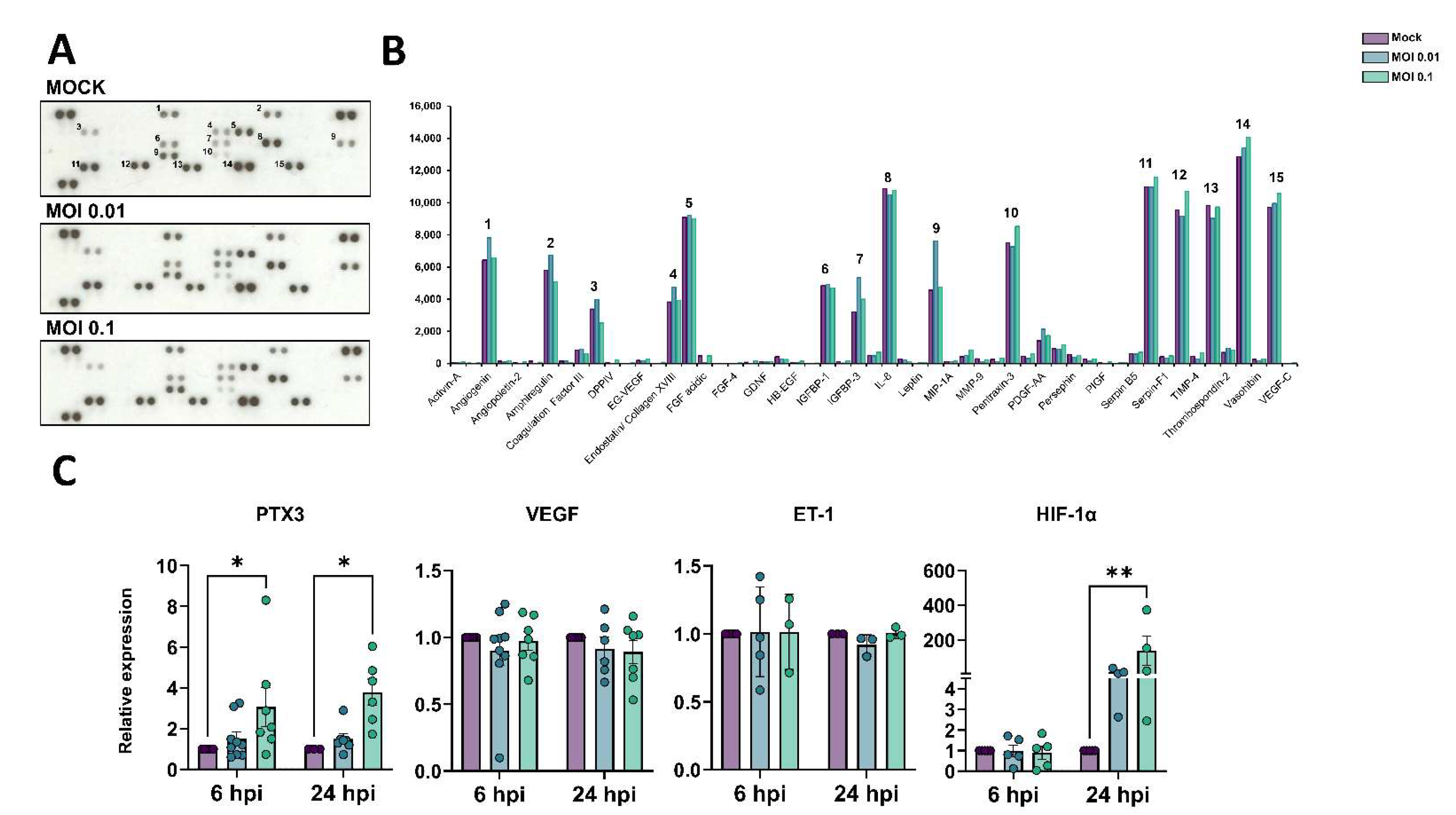
2.12. Angiogenesis-Related Protein Secretome
2.13. Transmission Electron Microscopy
2.14. Statistical Analyses
3. Results
3.1. Characterization of HBMEC Challenge by SARS-CoV-2
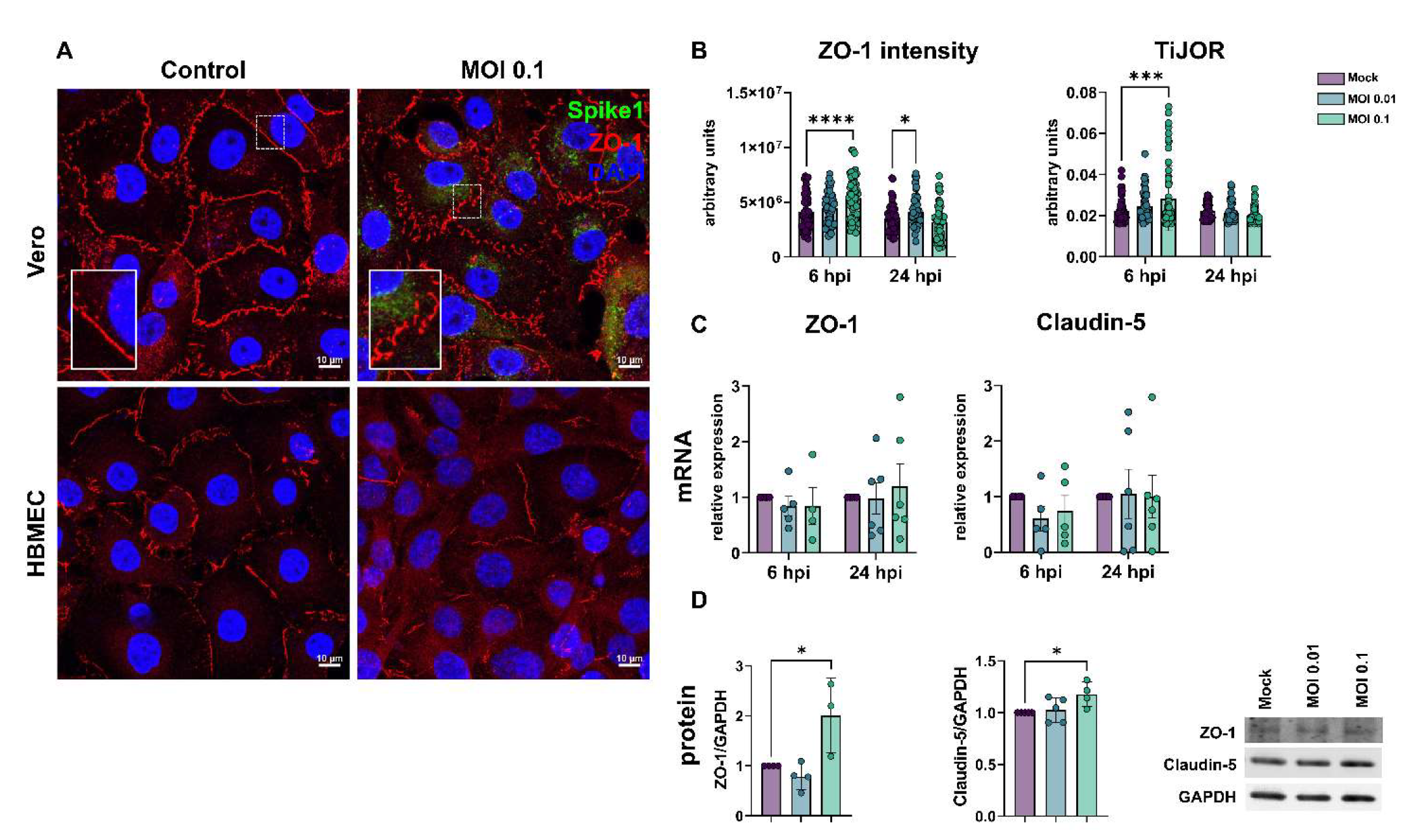
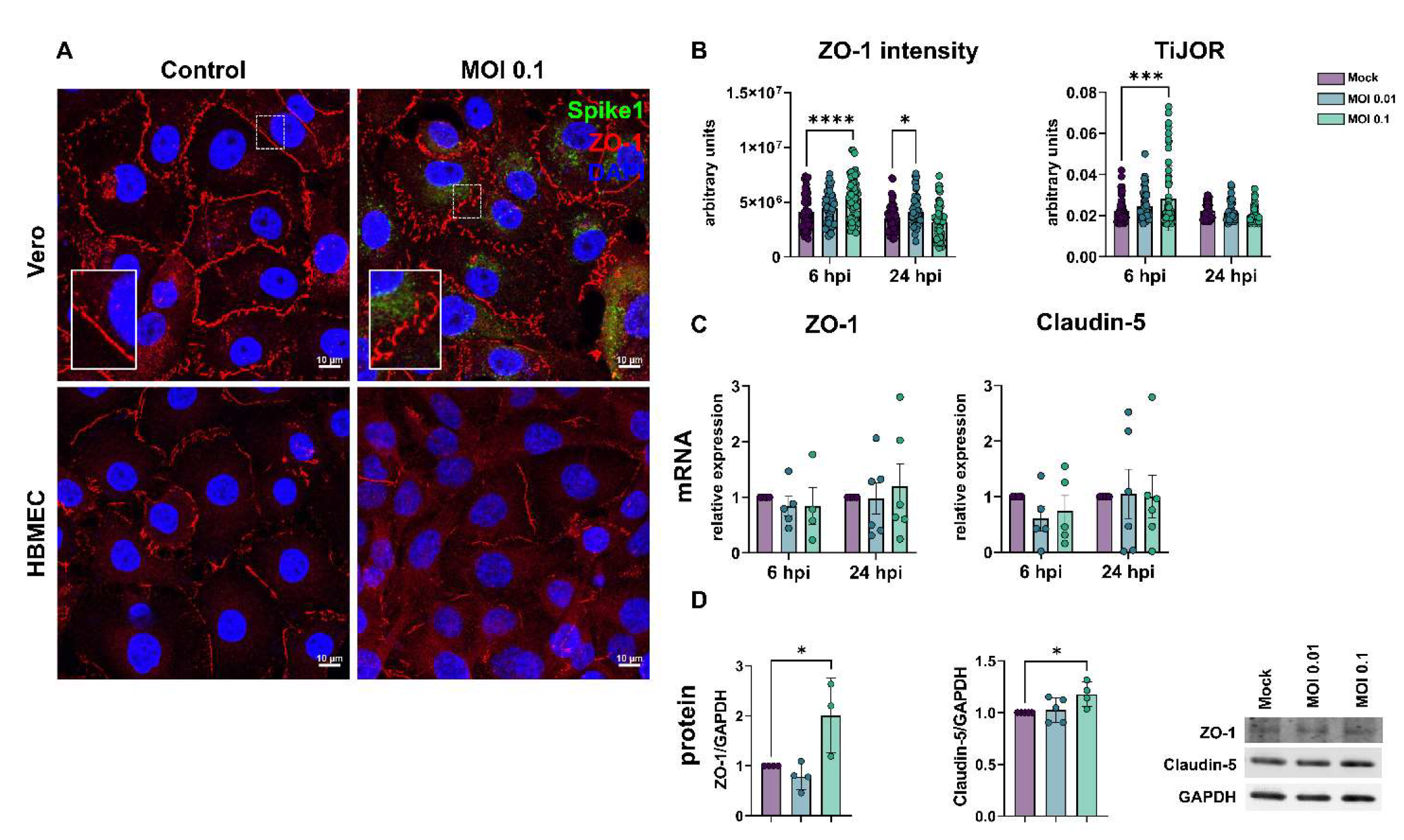
3.2. SARS-CoV-2 Affects Tight Junction Genes Expression in BBB-Forming Cells
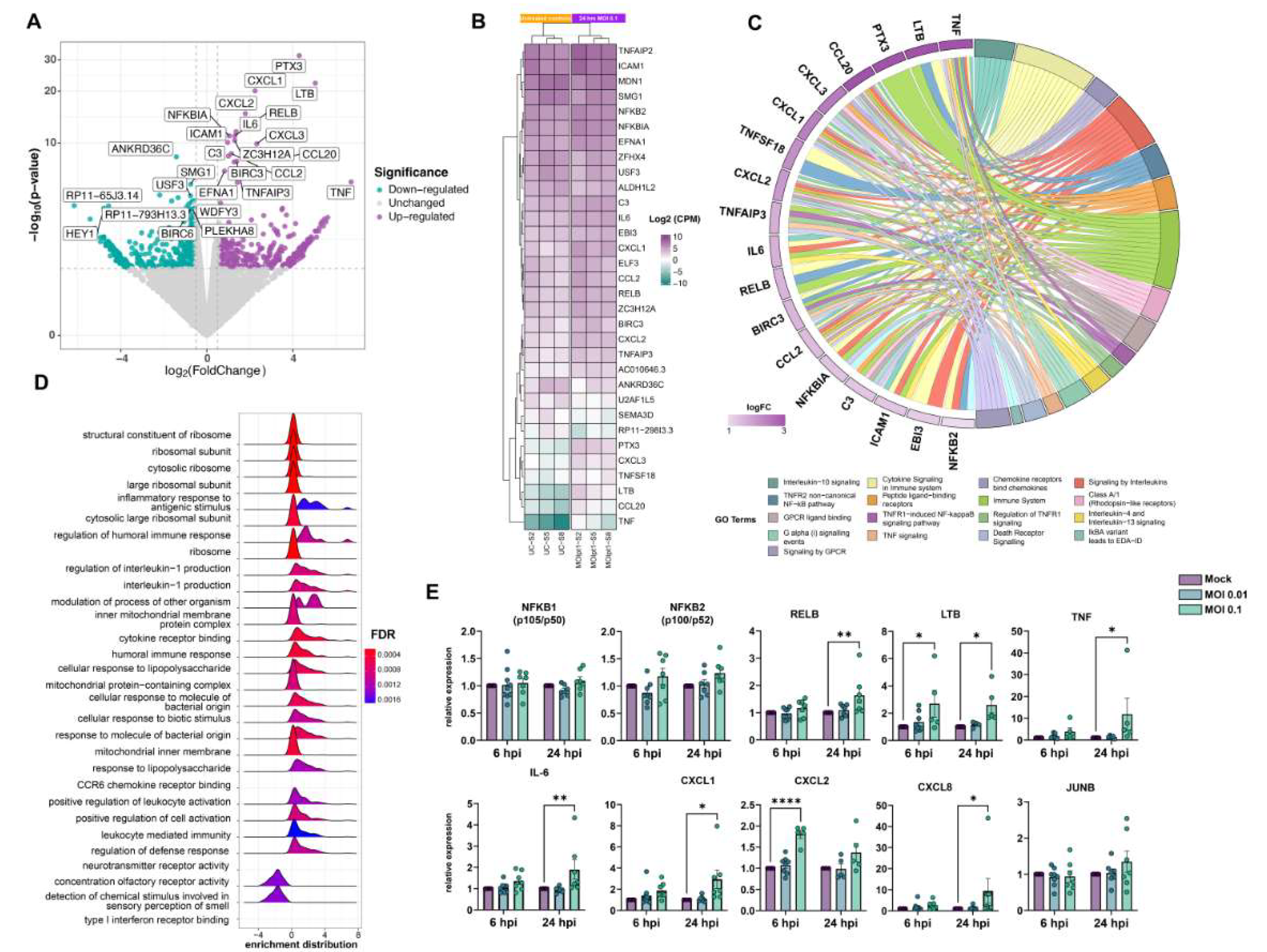
3.3. Exposure to SARS-CoV-2 Promotes Endothelial Activation and Hyper-Inflammatory Response In Vitro
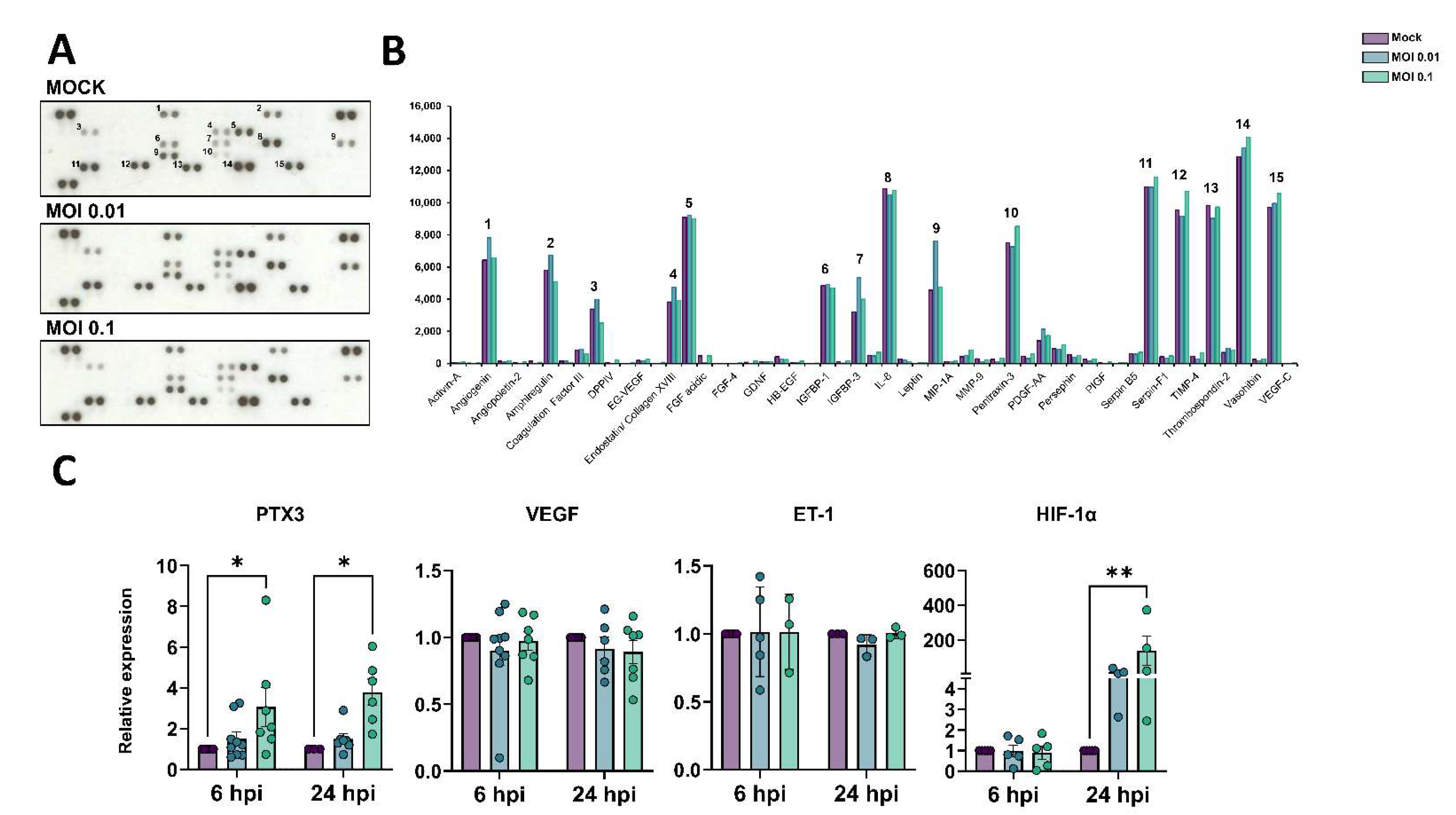
3.4. Angiogenic Profiling of SARS-CoV-2-Exposed HBMEC Cells
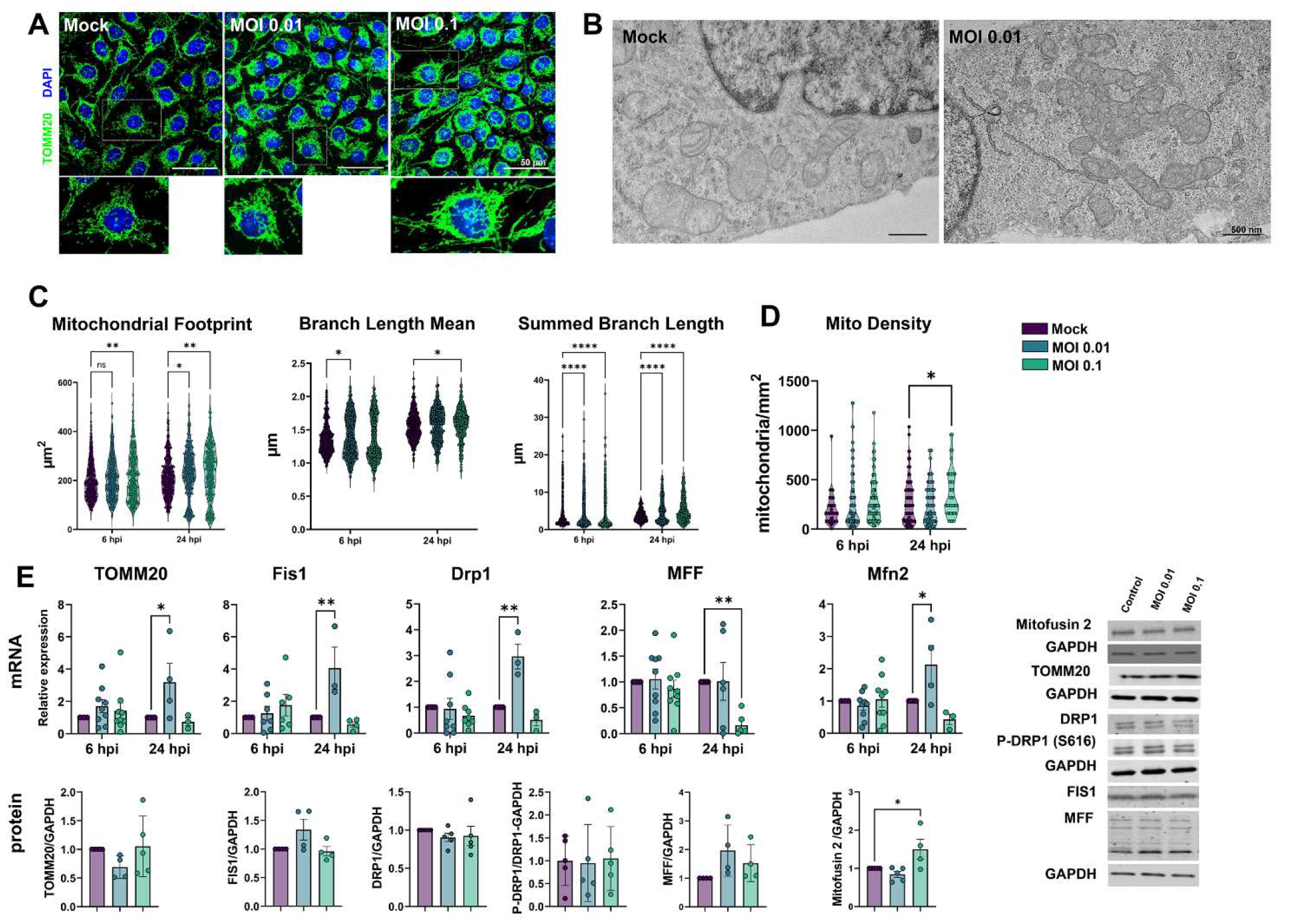
3.5. Mitochondrial Plasticity Is Affected by Exposure to SARS-CoV-2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Centers for Disease Control and Prevention (CDC). COVID Data Tracker. Available online: http://www.cdc.gov (accessed on 10 January 2023).
- WHO. Laboratory biosafety guidance related to coronavirus disease (COVID-19): Interim Guidance, 28 January 2021. 2021. Available online: https://www.who.int/publications/i/item/WHO-WPE-GIH-2021.1 (accessed on 10 January 2023).
- Centers for Disease Control and Prevention. Case-Surveillance. Available online: https://data.cdc.gov (accessed on 10 January 2023).
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Xiao, S.; Li, X.; Sun, L. ACE gene missense mutation in a case with early-onset, rapid progressing dementia. Gen. Psychiatry 2019, 32, e100028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torices, S.; Cabrera, R.; Stangis, M.; Naranjo, O.; Fattakhov, N.; Teglas, T.; Adesse, D.; Toborek, M. Expression of SARS-CoV-2-related receptors in cells of the neurovascular unit: Implications for HIV-1 infection. J. Neuroinflamm. 2021, 18, 67. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Zipeto, D.; Palmeira, J.D.F.; Arganaraz, G.A.; Arganaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [Google Scholar] [CrossRef]
- Schreiber, B.; Patel, A.; Verma, A. Shedding Light on COVID-19: ADAM17 the Missing Link. Am. J. Ther. 2020, 28, e358–e360. [Google Scholar] [CrossRef]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef]
- Bassendine, M.F.; Bridge, S.H.; McCaughan, G.W.; Gorrell, M.D. COVID-19 and comorbidities: A role for dipeptidyl peptidase 4 (DPP4) in disease severity. J. Diabetes 2020, 12, 649–658. [Google Scholar] [CrossRef]
- Cui, C.; Huang, C.; Zhou, W.; Ji, X.; Zhang, F.; Wang, L.; Zhou, Y.; Cui, Q. AGTR2, one possible novel key gene for the entry of SARS-CoV-2 into human cells. IEEE/ACM Trans Comput. Biol. Bioinform. 2020, 18, 1230–1233. [Google Scholar] [CrossRef]
- De Sousa, A.K.; Magalhaes, D.A.; Ferreira, J.D.S.; Barbosa, A. SARS-CoV-2-mediated encephalitis: Role of AT2R receptors in the blood-brain barrier. Med. Hypothes. 2020, 144, 110213. [Google Scholar] [CrossRef]
- Qiao, J.; Li, W.; Bao, J.; Peng, Q.; Wen, D.; Wang, J.; Sun, B. The expression of SARS-CoV-2 receptor ACE2 and CD147, and protease TMPRSS2 in human and mouse brain cells and mouse brain tissues. Biochem. Biophys. Res. Commun. 2020, 533, 867–871. [Google Scholar] [CrossRef]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Desikan, R.; Dixit, N.M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PloS Comput. Biol. 2020, 16, e1008461. [Google Scholar] [CrossRef]
- IstIfl, I.E.; SihoGlu Tepe, A.; SarikUrkc, U.C.; Tepe, B. Interaction of certain monoterpenoid hydrocarbons with the receptor binding domain of 2019 novel coronavirus (2019-nCoV), transmembrane serine protease 2 (TMPRSS2), cathepsin B, and cathepsin L (CatB/L) and their pharmacokinetic properties. Turk. J. Biol. 2020, 44, 242–264. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of COVID-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Virhammar, J.; Kumlien, E.; Fällmar, D.; Frithiof, R.; Jackmann, S.; Sköld, M.K.; Kadir, M.; Frick, J.; Lindeberg, J.; Olivero-Reinius, H.; et al. Acute necrotizing encephalopathy with SARS-CoV-2 RNA confirmed in cerebrospinal fluid. Neurology 2020, 95, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host–Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, M.; Wang, M.; Zhou, Y.; Chang, J.; Xian, Y.; Wang, D.; Mao, L.; Jin, H.; Hu, B. Acute cerebrovascular disease following COVID-19: A single center, retrospective, observational study. Stroke Vasc. Neurol. 2020, 5, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, T.; Yang, N.; Han, D.; Mi, X.; Li, Y.; Liu, K.; Vuylsteke, A.; Xiang, H.; Guo, X. Neurological manifestations of patients with COVID-19: Potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front. Med. 2020, 14, 533–541. [Google Scholar] [CrossRef]
- Barbosa, L.C.; Gonçalves, T.L.; de Araujo, L.P.; Rosario, L.V.D.O.; Ferrer, V.P. Endothelial cells and SARS-CoV-2: An intimate relationship. Vasc. Pharmacol. 2021, 137, 106829. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell. Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19). Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef]
- Stins, M.F.; Badgera, J.; Kim, K.S. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 2001, 30, 19–28. [Google Scholar] [CrossRef]
- WHO. Laboratory Biosafety Guidance Related to Coronavirus Disease (COVID-19): Interim Guidance. Available online: https://www.who.int/publications/i/item/WHO-WPE-GIH-2021.1 (accessed on 28 January 2021).
- Matos, A.D.R.; Caetano, B.C.; Filho, J.L.D.A.; Martins, J.S.C.D.C.; de Oliveira, M.G.P.; Sousa, T.d.C.; Horta, M.A.P.; Siqueira, M.M.; Fernandez, J.H. Identification of Hypericin as a Candidate Repurposed Therapeutic Agent for COVID-19 and Its Potential Anti-SARS-CoV-2 Activity. Front. Microbiol. 2022, 13, 828984. [Google Scholar] [CrossRef]
- Barreto-Vieira, D.F.; da Silva, M.A.N.; Garcia, C.C.; Miranda, M.D.; Matos, A.D.R.; Caetano, B.C.; Resende, P.C.; Motta, F.C.; Siqueira, M.M.; Girard-Dias, W.; et al. Morphology and morphogenesis of SARS-CoV-2 in Vero-E6 cells. Memórias Do Inst. Oswaldo Cruz 2021, 116, e200443. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. Clusterprofiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, 709. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, J.; Lee, S.; Park, M.; Kim, T.Y.; Park, M.G.; Choi, B.Y.; Kim, D.; Chang, H.; Kim, V.N.; Lee, V.N.K.A.C.J. Development of a Laboratory-safe and Low-cost Detection Protocol for SARS-CoV-2 of the Coronavirus Disease 2019 (COVID-19). Exp. Neurobiol. 2020, 29, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Arganda-Carreras, I.; Fernández-González, R.; Muñoz-Barrutia, A.; Ortiz-De-Solorzano, C. 3D reconstruction of histological sections: Application to mammary gland tissue. Microsc. Res. Tech. 2010, 73, 1019–1029. [Google Scholar] [CrossRef]
- Lee, T.; Kashyap, R.; Chu, C. Building Skeleton Models via 3-D Medial Surface Axis Thinning Algorithms. CVGIP Graph. Model. Image Process. 1994, 56, 462–478. [Google Scholar] [CrossRef]
- Terryn, C.; Sellami, M.; Fichel, C.; Diebold, M.-D.; Gangloff, S.; Le Naour, R.; Polette, M.; Zahm, J.-M. Rapid method of quantification of tight-junction organization using image analysis. Cytom. Part A 2012, 83A, 235–241. [Google Scholar] [CrossRef]
- Jiang, M.; Kolehmainen, P.; Kakkola, L.; Maljanen, S.; Melén, K.; Smura, T.; Julkunen, I.; Österlund, P. SARS-CoV-2 Isolates Show Impaired Replication in Human Immune Cells but Differential Ability to Replicate and Induce Innate Immunity in Lung Epithelial Cells. Microbiol. Spectr. 2021, 9, e0077421. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Mockenhaupt, K.; Gonsiewski, A.; Kordula, T. RelB and Neuroinflammation. Cells 2021, 10, 1609. [Google Scholar] [CrossRef]
- Presta, M.; Foglio, E.; Schuind, A.C.; Ronca, R. Long Pentraxin-3 Modulates the Angiogenic Activity of Fibroblast Growth Factor-2. Front. Immunol. 2018, 9, 2327. [Google Scholar] [CrossRef] [Green Version]
- Estato, V.; Stipursky, J.; Gomes, F.; Mergener, T.C.; Frazão-Teixeira, E.; Allodi, S.; Tibiriçá, E.; Barbosa, H.S.; Adesse, D. The Neurotropic Parasite Toxoplasma gondii Induces Sustained Neuroinflammation with Microvascular Dysfunction in Infected Mice. Am. J. Pathol. 2018, 188, 2674–2687. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK 1 phosphorylates Drp1 S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef]
- Sanchez, C.V.; Theel, E.; Binnicker, M.; Toledano, M.; McKeon, A. Autoimmune Encephalitis After SARS-CoV-2 Infection: Case Frequency, Findings, and Outcomes. Neurology 2021, 97, e2262–e2268. [Google Scholar] [CrossRef]
- Paterson, R.W.; Brown, R.L.; Benjamin, L.; Nortley, R.; Wiethoff, S.; Bharucha, T.; Jayaseelan, D.L.; Kumar, G.; Raftopoulos, R.E.; Zambreanu, L.; et al. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain 2020, 143, 3104–3120. [Google Scholar] [CrossRef]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van Den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mäe, M.; Muhl, L.; et al. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Conde, J.N.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. Mbio 2020, 11, e03185-20. [Google Scholar] [CrossRef]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef]
- Heuberger, J.; Trimpert, J.; Vladimirova, D.; Goosmann, C.; Lin, M.; Schmuck, R.; Mollenkopf, H.; Brinkmann, V.; Tacke, F.; Osterrieder, N.; et al. Epithelial response to IFN-γ promotes SARS-CoV-2 infection. EMBO Mol. Med. 2021, 13, e13191. [Google Scholar] [CrossRef] [PubMed]
- Meuren, L.M.; Prestes, E.B.; Papa, M.P.; de Carvalho, L.R.P.; Mustafá, Y.M.; da Costa, L.S.; Da Poian, A.T.; Bozza, M.T.; Arruda, L.B. Infection of Endothelial Cells by Dengue Virus Induces ROS Production by Different Sources Affecting Virus Replication, Cellular Activation, Death and Vascular Permeability. Front. Immunol. 2022, 13, 810376. [Google Scholar] [CrossRef] [PubMed]
- Leda, A.; Bertrand, L.; Andras, I.E.; El-Hage, N.; Nair, M.; Toborek, M. Selective Disruption of the Blood–Brain Barrier by Zika Virus. Front. Microbiol. 2019, 10, 2158. [Google Scholar] [CrossRef] [PubMed]
- Mladinich, M.C.; Conde, J.N.; Schutt, W.R.; Sohn, S.-Y.; Mackow, E.R. Blockade of Autocrine CCL5 Responses Inhibits Zika Virus Persistence and Spread in Human Brain Microvascular Endothelial Cells. Mbio 2021, 12, e01962-21. [Google Scholar] [CrossRef] [PubMed]
- DeOre, B.J.; Tran, K.A.; Andrews, A.M.; Ramirez, S.H.; Galie, P.A. SARS-CoV-2 Spike Protein Disrupts Blood–Brain Barrier Integrity via RhoA Activation. J. Neuroimmune Pharmacol. 2021, 16, 722–728. [Google Scholar] [CrossRef]
- Hao, S.; Ning, K.; Kuz, C.A.; Vorhies, K.; Yan, Z.; Qiu, J. Long-Term Modeling of SARS-CoV-2 Infection of In Vitro Cultured Polarized Human Airway Epithelium. Mbio 2020, 11, e02852-20. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A.; et al. Effect of SARS-CoV-2 proteins on vascular permeability. Elife 2021, 10, e69314. [Google Scholar] [CrossRef]
- Saras, J.; Heldin, C. PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem. Sci. 1996, 21, 455–458. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Bandyopadhyay, M. Molecular docking of SARS-CoV-2 Spike epitope sequences identifies heterodimeric peptide-protein complex formation with human Zo-1, TLR8 and brain specific glial proteins. Med. Hypotheses 2021, 157, 110706. [Google Scholar] [CrossRef]
- Caillet-Saguy, C.; Wolff, N. PDZ-Containing Proteins Targeted by the ACE2 Receptor. Viruses 2021, 13, 2281. [Google Scholar] [CrossRef]
- Feng, S.; Zou, L.; Wang, H.; He, R.; Liu, K.; Zhu, H. RhoA/ROCK-2 Pathway Inhibition and Tight Junction Protein Upregulation by Catalpol Suppresses Lipopolysaccaride-Induced Disruption of Blood-Brain Barrier Permeability. Molecules 2018, 23, 2371. [Google Scholar] [CrossRef] [Green Version]
- Mariano, C.; Palmela, I.; Pereira, P.; Fernandes, A.; Falcão, A.; Cardoso, F.L.; Vaz, A.R.; Campos, A.R.; Ferreira, A.J.D.C.G.; Kim, K.S.; et al. Tricellulin expression in brain endothelial and neural cells. Cell Tissue Res. 2012, 351, 397–407. [Google Scholar] [CrossRef]
- Constant, O.; Barthelemy, J.; Bolloré, K.; Tuaillon, E.; Gosselet, F.; Chable-Bessia, C.; Merida, P.; Muriaux, D.; Van de Perre, P.; Salinas, S.; et al. SARS-CoV-2 Poorly Replicates in Cells of the Human Blood-Brain Barrier Without Associated Deleterious Effects. Front. Immunol. 2021, 12, 697329. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood–brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 2021, 6, 337. [Google Scholar] [CrossRef]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan; et al. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Shih, V.F.-S.; Davis-Turak, J.; Macal, M.; Huang, J.Q.; Ponomarenko, J.; Kearns, J.D.; Yu, T.; Fagerlund, R.; Asagiri, M.; Zuniga, I.E.; et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat. Immunol. 2012, 13, 1162–1170. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.; McCall, C.E. NAD+-dependent SIRT1 Deacetylase Participates in Epigenetic Reprogramming during Endotoxin Tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; De Lott, L.B.; Nan, B.; Elner, V.M.; Sawalha, A.H. DNA methylation analysis of the temporal artery microenvironment in giant cell arteritis. Ann. Rheum. Dis. 2015, 75, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gorelik, G.; Strickland, F.M.; Richardson, B.C. Oxidative Stress, T Cell DNA Methylation, and Lupus. Arthritis Rheumatol. 2014, 66, 1574–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oaks, Z.; Perl, A. Metabolic control of the epigenome in systemic Lupus erythematosus. Autoimmunity 2013, 47, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.J.; Pang, A.P.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef]
- Mantovani, A.; Netea, M.G. Trained Innate Immunity, Epigenetics, and COVID-19. N. Engl. J. Med. 2020, 383, 1078–1080. [Google Scholar] [CrossRef]
- Ranjbar, M.; Rahimi, A.; Baghernejadan, Z.; Ghorbani, A.; Khorramdelazad, H. Role of CCL2/CCR2 axis in the pathogenesis of COVID-19 and possible Treatments: All options on the Table. Int. Immunopharmacol. 2022, 113, 109325. [Google Scholar] [CrossRef]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; Girndt, M.; et al. Liquid biomarkers of macrophage dysregulation and circulating spike protein illustrate the biological heterogeneity in patients with post-acute sequelae of COVID-19. J. Med. Virol. 2022, 95, e28364. [Google Scholar] [CrossRef]
- Gedda, M.R.; Danaher, P.; Shao, L.; Ongkeko, M.; Chen, L.; Dinh, A.; Sall, M.T.; Reddy, O.L.; Bailey, C.; Wahba, A.; et al. Longitudinal transcriptional analysis of peripheral blood leukocytes in COVID-19 convalescent donors. J. Transl. Med. 2022, 20, 587. [Google Scholar] [CrossRef]
- Amer, O.E.; Sabico, S.; Sheshah, E.; Alotaibi, N.H.; Aldisi, D.A.; Enani, M.A.; Aljohani, N.J.; Alshingetti, N.; Alomar, S.Y.; Hussain, S.D.; et al. Evaluation of 34 Cytokines and Vitamin D Status Reveal A Sexually-Dimorphic Active Immune Response to SARS-CoV-2. Healthcare 2022, 10, 2571. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Rafiullah, M.; Alkhowaiter, M.; Alotaibi, N.; Alzahrani, M.; Binkhamis, K.; Siddiqui, K.; Youssef, A.; Altalhi, H.; Almaghlouth, I.; et al. Clinical and biochemical characteristics of people experiencing post-coronavirus disease 2019-related symptoms: A prospective follow-up investigation. Front. Med. 2022, 9, 12. [Google Scholar] [CrossRef]
- Liu, K.K.; Dorovini-Zis, K. Differential Regulation of CD4+ T Cell Adhesion to Cerebral Microvascular Endothelium by the β-Chemokines CCL2 and CCL3. Int. J. Mol. Sci. 2012, 13, 16119–16140. [Google Scholar] [CrossRef] [Green Version]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/Monocyte Chemoattractant Protein-1 Mediates Enhanced Transmigration of Human Immunodeficiency Virus (HIV)-Infected Leukocytes across the Blood–Brain Barrier: A Potential Mechanism of HIV–CNS Invasion and NeuroAIDS. J. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, N.K.; Williams, R.; Callen, S.; Zien, C.; Narayan, O.; Buch, S. Roles of MCP-1 in development of HIV-dementia. Front. Biosci. 2008, 13, 3913–3918. [Google Scholar] [CrossRef] [Green Version]
- Daigo, K.; Mantovani, A.; Bottazzi, B. The yin-yang of long pentraxin PTX3 in inflammation and immunity. Immunol. Lett. 2014, 161, 38–43. [Google Scholar] [CrossRef]
- Muffley, L.A.; Pan, S.-C.; Smith, A.N.; Ga, M.; Hocking, A.M.; Gibran, N.S. Differentiation state determines neural effects on microvascular endothelial cells. Exp. Cell Res. 2012, 318, 2085–2093. [Google Scholar] [CrossRef] [Green Version]
- Freese, C.; Hanada, S.; Fallier-Becker, P.; Kirkpatrick, C.J.; Unger, R.E. Identification of neuronal and angiogenic growth factors in an in vitro blood-brain barrier model system: Relevance in barrier integrity and tight junction formation and complexity. Microvasc. Res. 2017, 111, 1–11. [Google Scholar] [CrossRef]
- Shindo, A.; Maki, T.; Mandeville, E.T.; Liang, A.C.; Egawa, N.; Itoh, K.; Itoh, N.; Borlongan, M.; Holder, J.C.; Chuang, T.T.; et al. Astrocyte-Derived Pentraxin 3 Supports Blood–Brain Barrier Integrity Under Acute Phase of Stroke. Stroke 2016, 47, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Wesley, U.V.; Sutton, I.; Clark, P.A.; Cunningham, K.; Larrain, C.; Kuo, J.S.; Dempsey, R.J. Enhanced expression of pentraxin-3 in glioblastoma cells correlates with increased invasion and IL8-VEGF signaling axis. Brain Res. 2021, 1776, 147752. [Google Scholar] [CrossRef]
- Siqueira, M.; Francis, D.; Gisbert, D.; Gomes, F.C.A.; Stipursky, J. Radial Glia Cells Control Angiogenesis in the Developing Cerebral Cortex Through TGF-β1 Signaling. Mol. Neurobiol. 2017, 55, 3660–3675. [Google Scholar] [CrossRef]
- Breviario, F.; D’Aniello, E.M.; Golay, J.T.; Peri, G.; Bottazzi, B.; Bairoch, A.; Saccone, S.; Marzella, R.; Predazzi, V.; Rocchi, M. Interleukin-1-inducible genes in endothelial cells. Cloning of a new gene related to C-reactive protein and serum amyloid P component. J. Biol. Chem. 1992, 267, 22190–22197. [Google Scholar] [CrossRef] [PubMed]
- Fazzini, F.; Peri, G.; Doni, A.; Dell’Antonio, G.; Cin, E.D.; Bozzolo, E.; D’Auria, F.; Praderio, L.; Ciboddo, G.; Sabbadini, M.G.; et al. PTX3 in small-vessel vasculitides: An independent indicator of disease activity produced at sites of inflammation. Arthritis Rheum. 2001, 44, 2841–2850. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Camozzi, M.; Moroni, E.; Bottazzi, B.; Peri, G.; Indraccolo, S.; Amadori, A.; Mantovani, A.; Presta, M. Selective recognition of fibroblast growth factor-2 by the long pentraxin PTX3 inhibits angiogenesis. Blood 2004, 104, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meini, S.; Giani, T.; Tascini, C. Intussusceptive angiogenesis in COVID-19: Hypothesis on the significance and focus on the possible role of FGF2. Mol. Biol. Rep. 2020, 47, 8301–8304. [Google Scholar] [CrossRef]
- Ackermann, M.; Mentzer, S.J.; Kolb, M.; Jonigk, D. Inflammation and intussusceptive angiogenesis in COVID-19: Everything in and out of flow. Eur. Respir. J. 2020, 56, 2003147. [Google Scholar] [CrossRef]
- Mentzer, S.J.; Ackermann, M.; Jonigk, D. Endothelialitis, Microischemia, and Intussusceptive Angiogenesis in COVID-19. Cold Spring Harb. Perspect. Med. 2022, 12, a041157. [Google Scholar] [CrossRef]
- Maldonado, F.; Morales, D.; Díaz-Papapietro, C.; Valdés, C.; Fernandez, C.; Valls, N.; Lazo, M.; Espinoza, C.; González, R.; Gutiérrez, R.; et al. Relationship Between Endothelial and Angiogenesis Biomarkers Envisage Mortality in a Prospective Cohort of COVID-19 Patients Requiring Respiratory Support. Front. Med. 2022, 9, 826218. [Google Scholar] [CrossRef]
- Gopinathan, G.; Milagre, C.; Pearce, O.M.; Reynolds, L.E.; Hodivala-Dilke, K.; Leinster, D.A.; Zhong, H.; Hollingsworth, R.E.; Thompson, R.; Whiteford, J.R.; et al. Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res. 2015, 75, 3098–3107. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Corrales, N.; Gálvez, N.M.S.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Contribution of hypoxia inducible factor-1 during viral infections. Virulence 2020, 11, 1482–1500. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Afsar, B.; Kanbay, M.; Afsar, R.E. Hypoxia inducible factor-1 protects against COVID-19: A hypothesis. Med. Hypotheses 2020, 143, 109857. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Imperio, G.E.; Lye, P.; Mughis, H.; Hamada, H.; Bloise, E.; Lye, S.J.; Matthews, S.G. Hypoxia alters the expression of ACE2 and TMPRSS2 SARS-CoV-2 cell entry mediators in hCMEC/D3 brain endothelial cells. Microvasc. Res. 2021, 138, 104232. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxiainduced neutrophil survival is mediated by HIF-1αdependent NF-κB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Parodi-Rullán, R.M.; Javadov, S.; Fossati, S. Dissecting the Crosstalk between Endothelial Mitochondrial Damage, Vascular Inflammation, and Neurodegeneration in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Cells 2021, 10, 2903. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial Crisis in Cerebrovascular Endothelial Cells Opens the Blood–Brain Barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Romão, P.R.T.; Teixeira, P.C.; Schipper, L.; da Silva, I.; Filho, P.S.; Júnior, L.C.R.; Peres, A.; da Fonseca, S.G.; Monteiro, M.C.; Lira, F.S.; et al. Viral load is associated with mitochondrial dysfunction and altered monocyte phenotype in acute severe SARS-CoV-2 infection. Int. Immunopharmacol. 2022, 108, 108697. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Di Domizio, J.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Preston, K.J.; Cooper, H.A.; Boyer, M.J.; Escoto, K.M.; Poltronetti, A.J.; Elliott, K.J.; Kuroda, R.; Miyao, M.; Sesaki, H.; et al. Mitochondrial Fission Mediates Endothelial Inflammation. Hypertension 2020, 76, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Jendrach, M.; Pohl, S.; Vöth, M.; Kowald, A.; Hammerstein, P.; Bereiter-Hahn, J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, E.M.; Kruckeberg, T.W.; Comerford, L.E.; Buschmann, M.T. Thinning of Capillary Walls and Declining Numbers of Endothelial Mitochondria in the Cerebral Cortex of the Aging Primate, Macaca Nemestrina. J. Gerontol. 1979, 34, 642–650. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, Y.; Liu, J.; Liu, K.; Wen, J.; Wen, S.; Wu, Z. HSG/Mfn2 Gene Polymorphism and Essential Hypertension: A Case-Control Association Study in Chinese. J. Atheroscler. Thromb. 2011, 18, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta BBA Bioenerg. 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225, Erratum in Nature 2011, 475, 122. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Symbol | Fold-Change (Log) | p Value | FDR |
|---|---|---|---|---|
| Uncategorized gene | RP11-298I3.3 | −2.19 | 0.00001795234598 | 0.04214061886 |
| ankyrin repeat domain 36C | ANKRD36C | −1.42 | 0.000000005528613831 | 0.00002495701339 |
| upstream transcription factor family member 3 | USF3 | −0.76 | 0.00002036226065 | 0.04595918861 |
| SMG1 nonsense mediated mRNA decay associated PI3K related kinase | SMG1 | −0.75 | 0.000002651386726 | 0.007779698933 |
| nuclear factor kappa B subunit 2 | NFKB2 | 0.64 | 0.00002117775188 | 0.04602945153 |
| E74 like ETS transcription factor 3 | ELF3 | 0.82 | 0.00001027948854 | 0.02601045479 |
| ephrin A1 | EFNA1 | 0.82 | 0.0000001766175517 | 0.0006096837885 |
| Epstein–Barr-virus-induced 3 | EBI3 | 0.87 | 0.000005771957935 | 0.01612959902 |
| intercellular adhesion molecule 1 | ICAM1 | 0.97 | 0 | 0.0000005593411357 |
| complement C3 | C3 | 1.01 | 0.000000004506547278 | 0.00002203851837 |
| NFKB inhibitor alpha | NFKBIA | 1.05 | 0 | 0.00000008288304584 |
| C-C motif chemokine ligand 2 | CCL2 | 1.14 | 0.000000002315586268 | 0.00001235344223 |
| baculoviral IAP repeat containing 3 | BIRC3 | 1.27 | 0.00000002052506134 | 0.00007528079373 |
| zinc finger CCCH-type containing 12A | ZC3H12A | 1.29 | 0 | 0.0000003145723235 |
| RELB proto-oncogene, NF-kB subunit | RELB | 1.30 | 0 | 0.00000008288304584 |
| Uncharacterized protein | AC010646.3 | 1.31 | 0.0000103901062 | 0.02601045479 |
| interleukin 6 | IL6 | 1.36 | 0 | 0.00000002581261588 |
| TNF alpha induced protein 3 | TNFAIP3 | 1.38 | 0.00000001761165902 | 0.00006890150651 |
| TNF alpha induced protein 2 | TNFAIP2 | 1.42 | 0.000001921435199 | 0.005934605433 |
| C-X-C motif chemokine ligand 2 | CXCL2 | 1.79 | 0 | 0 |
| TNF superfamily member 18 | TNFSF18 | 1.95 | 0.0000106374977 | 0.02601045479 |
| C-X-C motif chemokine ligand 1 | CXCL1 | 2.24 | 0 | 0 |
| C-X-C motif chemokine ligand 3 | CXCL3 | 2.32 | 0.0000000001312489102 | 0.0000007702211048 |
| C-C motif chemokine ligand 20 | CCL20 | 3.50 | 0.000000007991169552 | 0.00003349669957 |
| pentraxin 3 | PTX3 | 4.30 | 0 | 0 |
| lymphotoxin beta | LTB | 5.04 | 0 | 0 |
| Analyte Name (Alternate Nomenclature) | Gene ID | Foreground Pixel Intensity (Fold Change Relative to Mock) | ||
|---|---|---|---|---|
| Mock | MOI 0.01 | MOI 0.1 | ||
| Activin-A | 3624 | 55.5 | 32.5 (0.6) | 88.5 (1.63) |
| ADAMTS-1 | 9510 | 25.5 | 12.5 (0.52) | 45.5 (1.88) |
| Angiogenin (ANG) | 283 | 6427 | 7818 (1.21) | 6553 (1.01) |
| Angiopoietin-1 (Ang-1) | 284 | 147 | 86 (0.59) | 155 (1.07) |
| Angiopoietin-2 (Ang-2) | 285 | 39.5 | 12 (0.32) | 79.5 (2.08) |
| Angiostatin/ Plasminigen | 5340 | 144.5 | 10 (0.07) | 74 (0.52) |
| Amphiregulin (AR) | 374 | 5797 | 6744 (1.16) | 5064.5 (0.87) |
| Artemin | 9048 | 144.5 | 143 (0.99) | 64 (0.46) |
| Coagulation Factor III (TF) | 2152 | 818 | 887 (1.08) | 622 (0.76) |
| CXCL16 | 58,191 | 3387.5 | 3976.5 (1.17) | 2521.5 (0.74) |
| DPPIV (CD26) | 1803 | 72.5 | 1.5 (0.03) | 203 (2.83) |
| EGF | 1950 | 16.5 | 18 (1.15) | 40.5 (2.60) |
| EG-VEGF (PK1) | 84,432 | 188.5 | 164 (0.88) | 257 (1.38) |
| Endoglin (CD105) | 2022 | 17.5 | 17.5 (1.05) | 74.5 (4.40) |
| Endostatin/ Collagen XVIII | 80,781 | 3827 | 4747.5 (1.24) | 3931.5 (1.02) |
| Endothelin-1 (ET-1) | 1906 | 9087.5 | 9213 (1.01) | 9007.5 (0.99) |
| FGF acidic (FGF-1) | 2246 | 475 | 69.5 (0.15) | 472.5 (1) |
| FGF basic (FGF2) | 2247 | 16.5 | 5 (0.36) | 4.5 (0.42) |
| FGF-4 | 2249 | 0.5 | −1 | 36.5 (78.0) |
| FGF-7 (KGF) | 2252 | 77.5 | 15.5 (0.21) | 180 (2.34) |
| GDNF | 2668 | 101 | 82 (0.82) | 105 (1.06) |
| GM-CSF | 1437 | 419.5 | 275 (0.65) | 241.5 (0.58) |
| HB-EGF | 1839 | 71 | 32.5 (0.47) | 134 (1.92) |
| HGF | 3082 | 10.5 | 11 (1.14) | 25.5 (2.66) |
| IGFBP-1 | 3484 | 4841.5 | 4901 (1.01) | 4684.5 (0.96) |
| IGFBP-2 | 3485 | 118 | 22.5 (0.19) | 154.5 (1.33) |
| IGFBP-3 | 3486 | 3203 | 5335.5 (1.66) | 3991 (1.24) |
| IL-1β (IL-1F2) | 3553 | 512 | 470.5 (0.92) | 695.5 (1.36). |
| IL-8 (CXCL8) | 3576 | 10,875.5 | 10,484.5 (0.96) | 10,744 (0.98) |
| LAP (TGF B1) | 7040 | 292.5 | 219.5 (0.75) | 102 (0.35) |
| Leptin | 3952 | 1 | 44 (45.0) | 43.5 (46.0) |
| MCP-1 (CCL2) | 6347 | 4549 | 7624 (1.67) | 4759 (1.04) |
| MIP-1A (CCL3) | 6348 | 123 | 103 (0.84) | 149 (1.23) |
| MMP-8 | 4317 | 418 | 510.5 (1.22) | 841.5 (2.01) |
| MMP-9 | 4318 | 298.5 | 106.5 (0.36) | 206 (0.69) |
| NRG1-B1 (HRG1-β1) | 3084 | 256 | 107.5 (0.42) | 320 (1.25) |
| Pentraxin-3 (TSG-14) | 5806 | 7509.5 | 7263.5 (0.96) | 8523 (1.13) |
| PD-ECGF | 1890 | 441 | 310 (0.70) | 600 (1.36) |
| PDGF-AA | 5154 | 1411.5 | 2129.5 (1.50) | 1736.5 (1.23) |
| PDGF-AB/ PDGF-BB | 5155 | 923.5 | 878 (0.95) | 1161 (1.25) |
| Persephin | 5623 | 565.5 | 375.5 (0.66) | 490.5 (0.87) |
| Platelet Factor 4 (CXCL4) | 5196 | 261.5 | 150.5 (0.58) | 283.5 (1.09) |
| PIGF | 5228 | 50.5 | 14 (0.29) | 90 (1.83) |
| Prolactin | 5617 | 7 | 49 (7.14) | 57 (8.5) |
| Serpin B5 (Maspin) | 5268 | 604 | 578 (0.95) | 694 (1.15) |
| Serpin-E1 (PAI-1) | 5054 | 10,979.5 | 10,955.5 (0.99) | 11,590.5 (1.05) |
| Serpin-F1 (PEDF) | 5176 | 422.5 | 327.5 (0.77) | 476 (1.13) |
| TIMP-1 | 7076 | 9548 | 9156 (0.95) | 10,696 (1.12) |
| TIMP-4 | 7079 | 446.5 | 292.5 (0.65) | 656.5 (1.47) |
| Thrombospondin-1 (TSP-1) | 7057 | 9842 | 9012 (0.91) | 9727.5 (0.98) |
| Thrombospondin-2 (TSP-2) | 7058 | 688 | 929 (1.35) | 837 (1.22) |
| uPA | 5328 | 12,865 | 13,396.5 (1.04) | 14,065 (1.09) |
| Vasohibin | 22,846 | 253 | 172 (0.68) | 268 (1.06) |
| VEGF | 7422 | 9700.5 | 9954 (10.2) | 10,559.5 (1.08) |
| VEGF-C | 7424 | 0.5 | 0 | 37 (79.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motta, C.S.; Torices, S.; da Rosa, B.G.; Marcos, A.C.; Alvarez-Rosa, L.; Siqueira, M.; Moreno-Rodriguez, T.; Matos, A.d.R.; Caetano, B.C.; Martins, J.S.C.d.C.; et al. Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling. Viruses 2023, 15, 745. https://doi.org/10.3390/v15030745
Motta CS, Torices S, da Rosa BG, Marcos AC, Alvarez-Rosa L, Siqueira M, Moreno-Rodriguez T, Matos AdR, Caetano BC, Martins JSCdC, et al. Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling. Viruses. 2023; 15(3):745. https://doi.org/10.3390/v15030745
Chicago/Turabian StyleMotta, Carolline Soares, Silvia Torices, Barbara Gomes da Rosa, Anne Caroline Marcos, Liandra Alvarez-Rosa, Michele Siqueira, Thaidy Moreno-Rodriguez, Aline da Rocha Matos, Braulia Costa Caetano, Jessica Santa Cruz de Carvalho Martins, and et al. 2023. "Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling" Viruses 15, no. 3: 745. https://doi.org/10.3390/v15030745
APA StyleMotta, C. S., Torices, S., da Rosa, B. G., Marcos, A. C., Alvarez-Rosa, L., Siqueira, M., Moreno-Rodriguez, T., Matos, A. d. R., Caetano, B. C., Martins, J. S. C. d. C., Gladulich, L., Loiola, E., Bagshaw, O. R. M., Stuart, J. A., Siqueira, M. M., Stipursky, J., Toborek, M., & Adesse, D. (2023). Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling. Viruses, 15(3), 745. https://doi.org/10.3390/v15030745