
An Endogenous Retrovirus Vaccine Encoding an Envelope with a Mutated Immunosuppressive Domain in Combination with Anti-PD1 Treatment Eradicates Established Tumours in Mice
 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Adenoviral Vaccine Vectors
2.2. Cell Culture
2.3. Mice
2.4. Mouse BMDCs
2.5. Viral Transduction
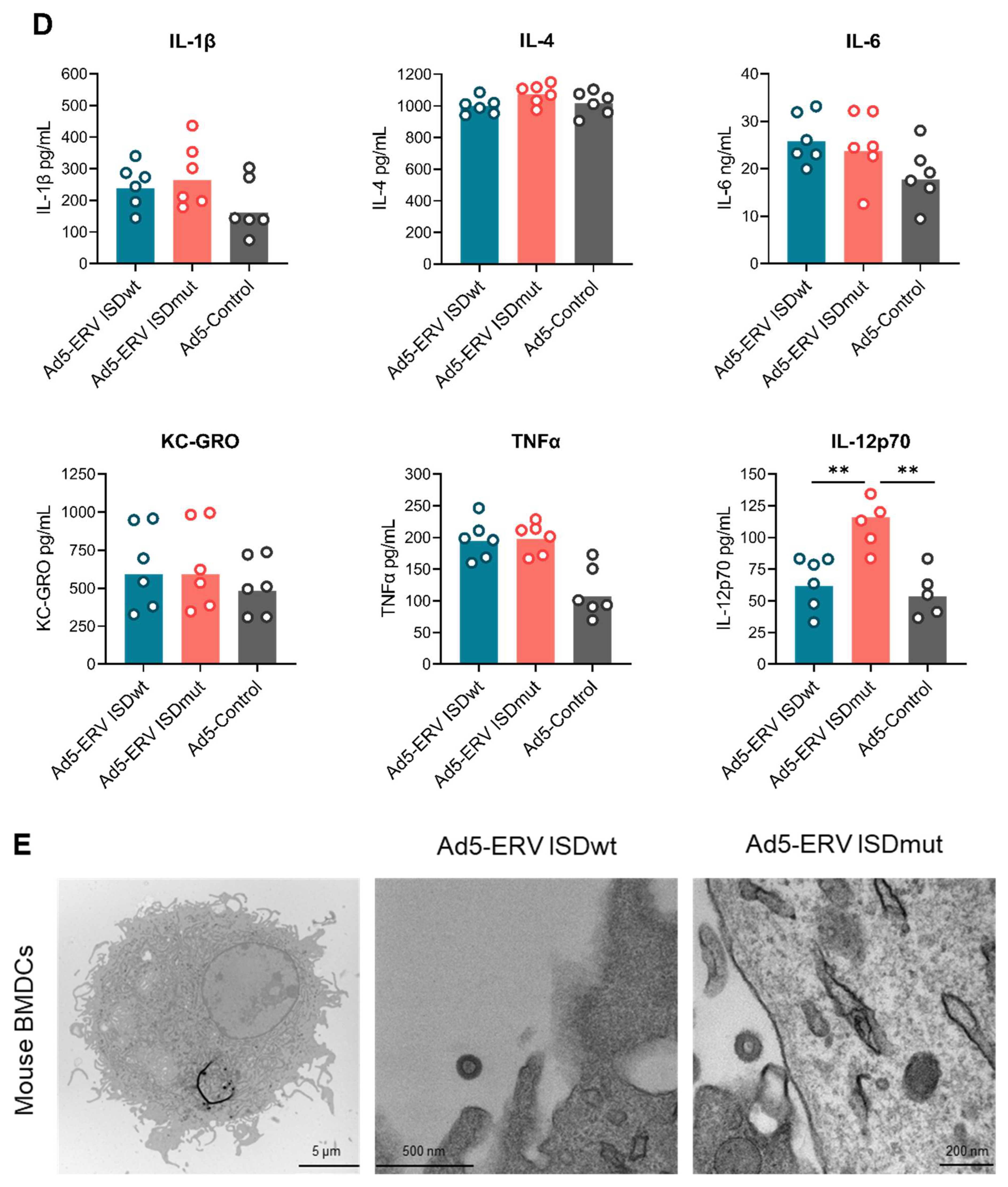
2.6. Transmission Electron Microscopy
2.7. Single Cell RNA Sequencing and Analysis
2.8. Western Blot
2.9. Staining for Flow Cytometry
2.10. Cytokine Screening of Transduced ERV Mouse BMDCs
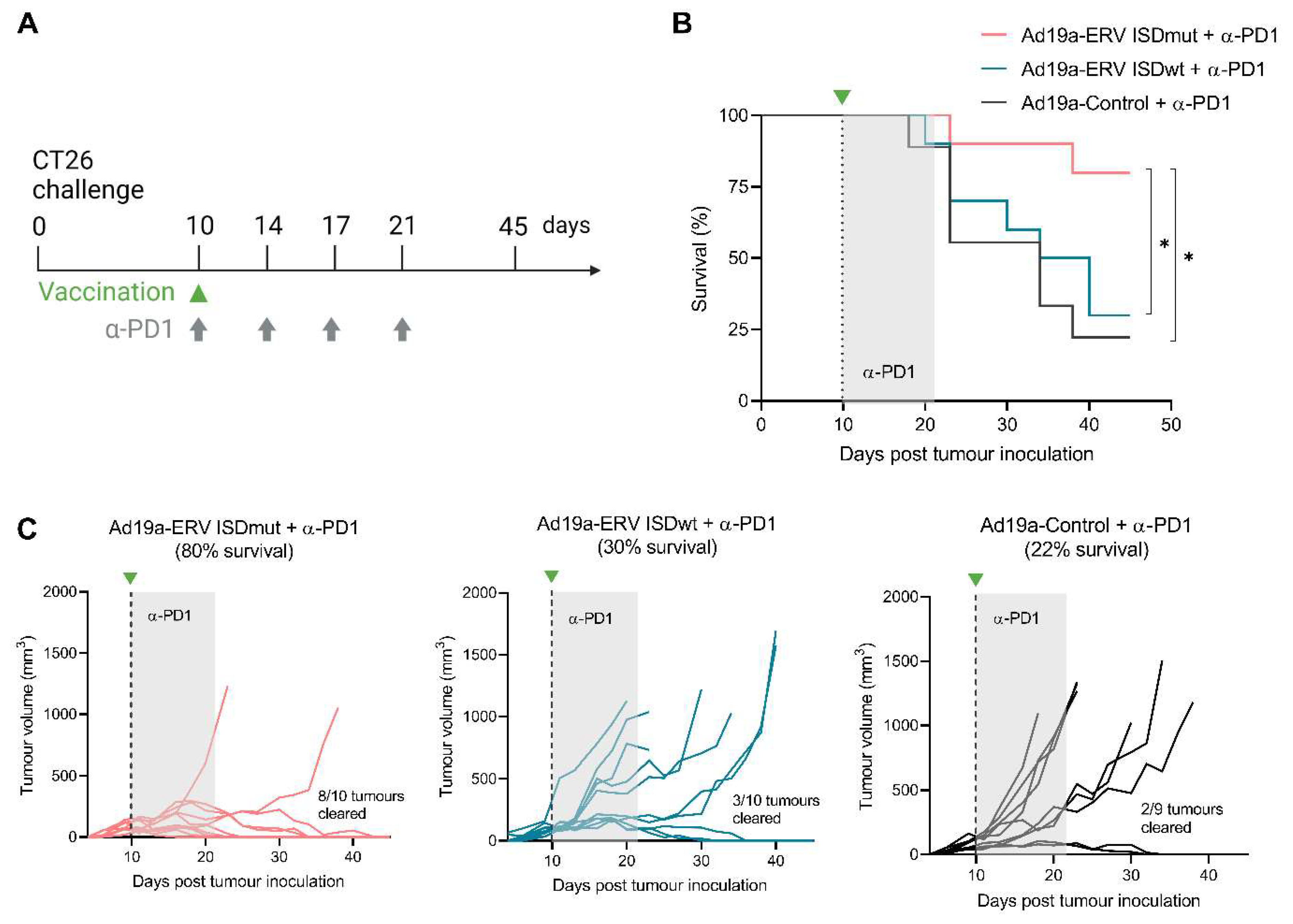
2.11. Tumour Challenge
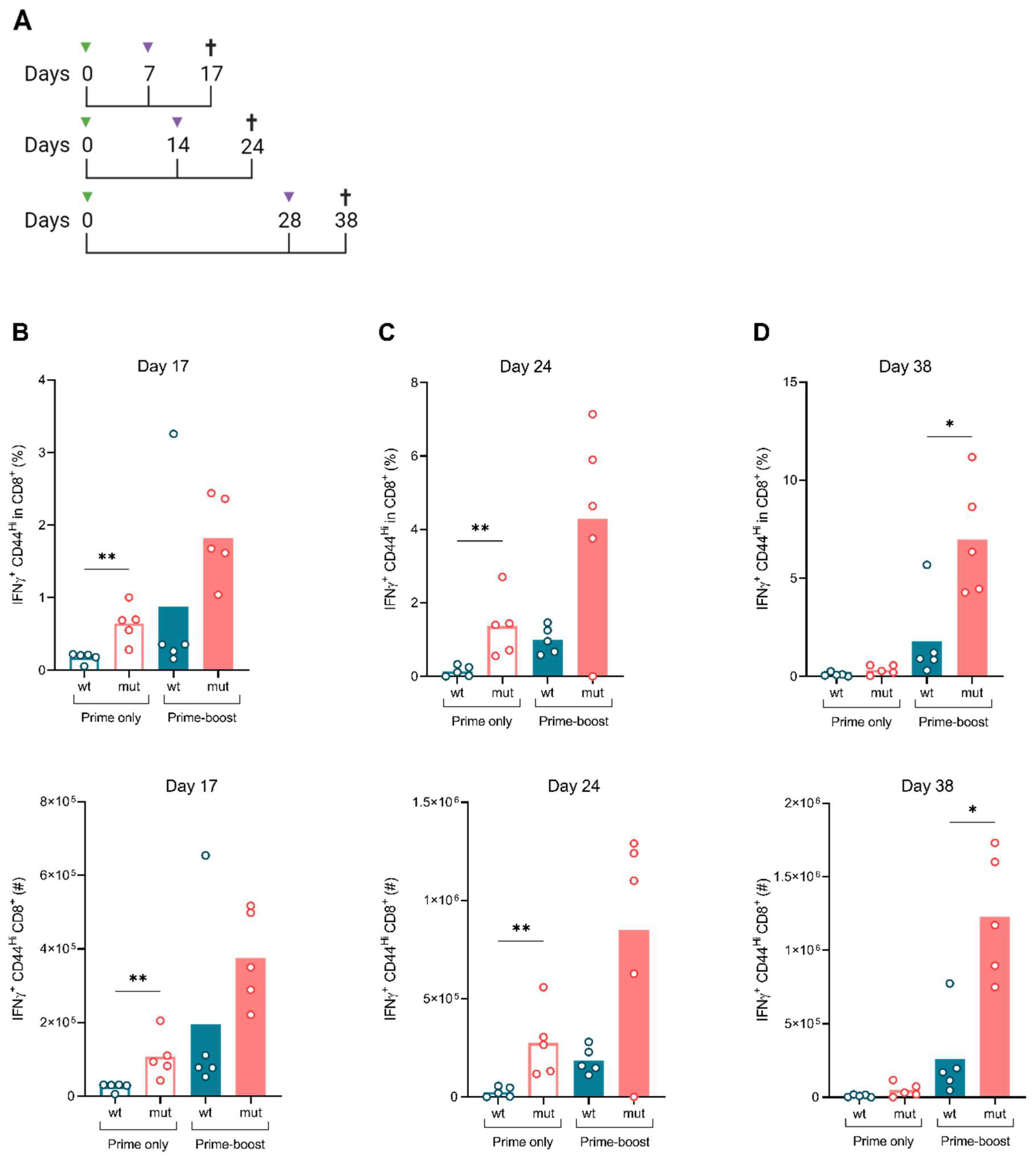
2.12. Immunization and Treatment
2.13. Enzyme Linked Immunosorbent Assay (ELISA)
2.14. Statistical Analyses
3. Results
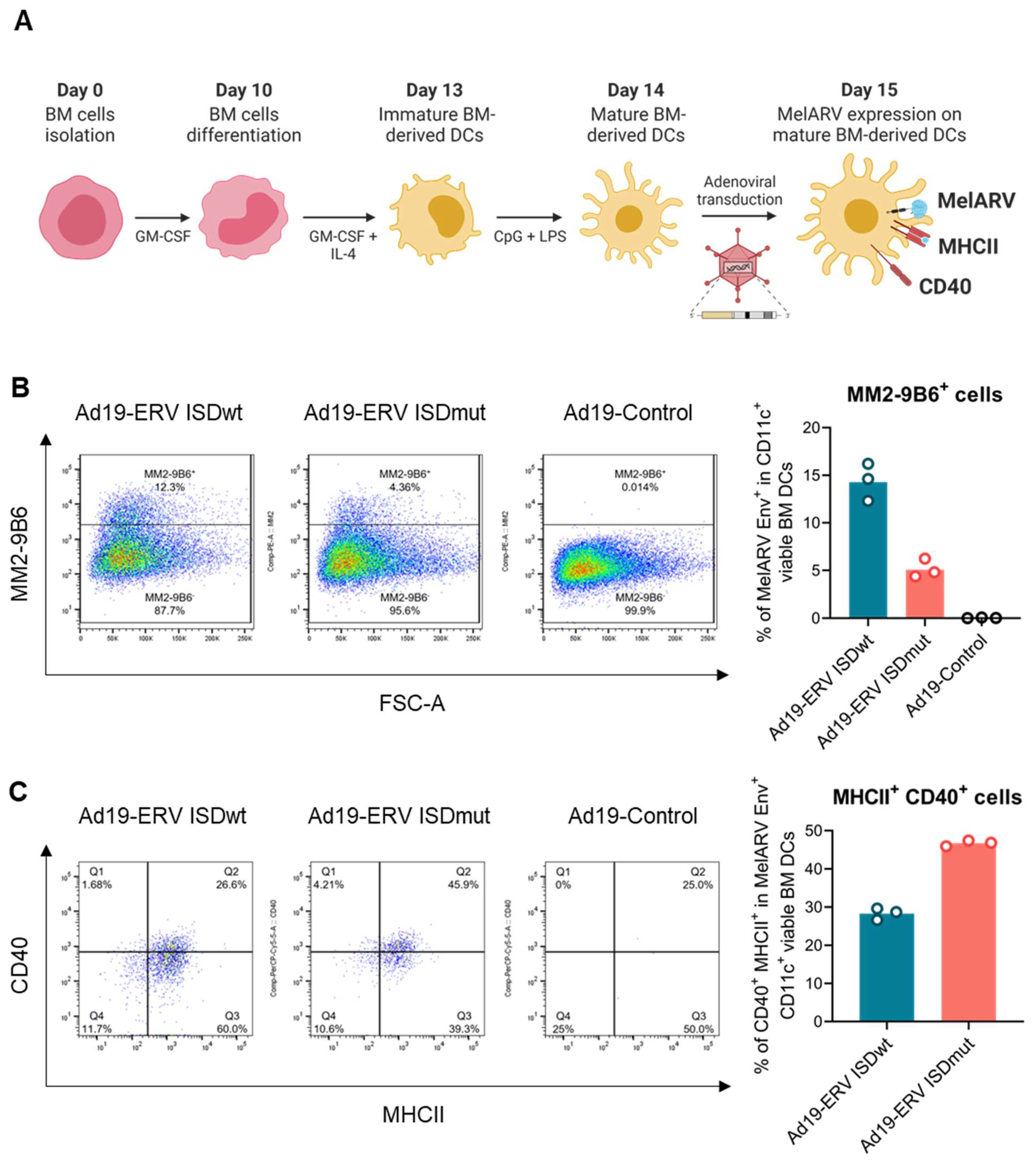
3.1. Characterization of the Virus-like Vaccines
3.2. ISDmut Vaccine Increased Mouse BMDC Activation
3.3. ISDmut Increased Vaccine Induced T Cell Responses
3.4. ISDmut Vaccine Synergized with α-PD1 and Eradicated Established Colorectal Tumours in Mice
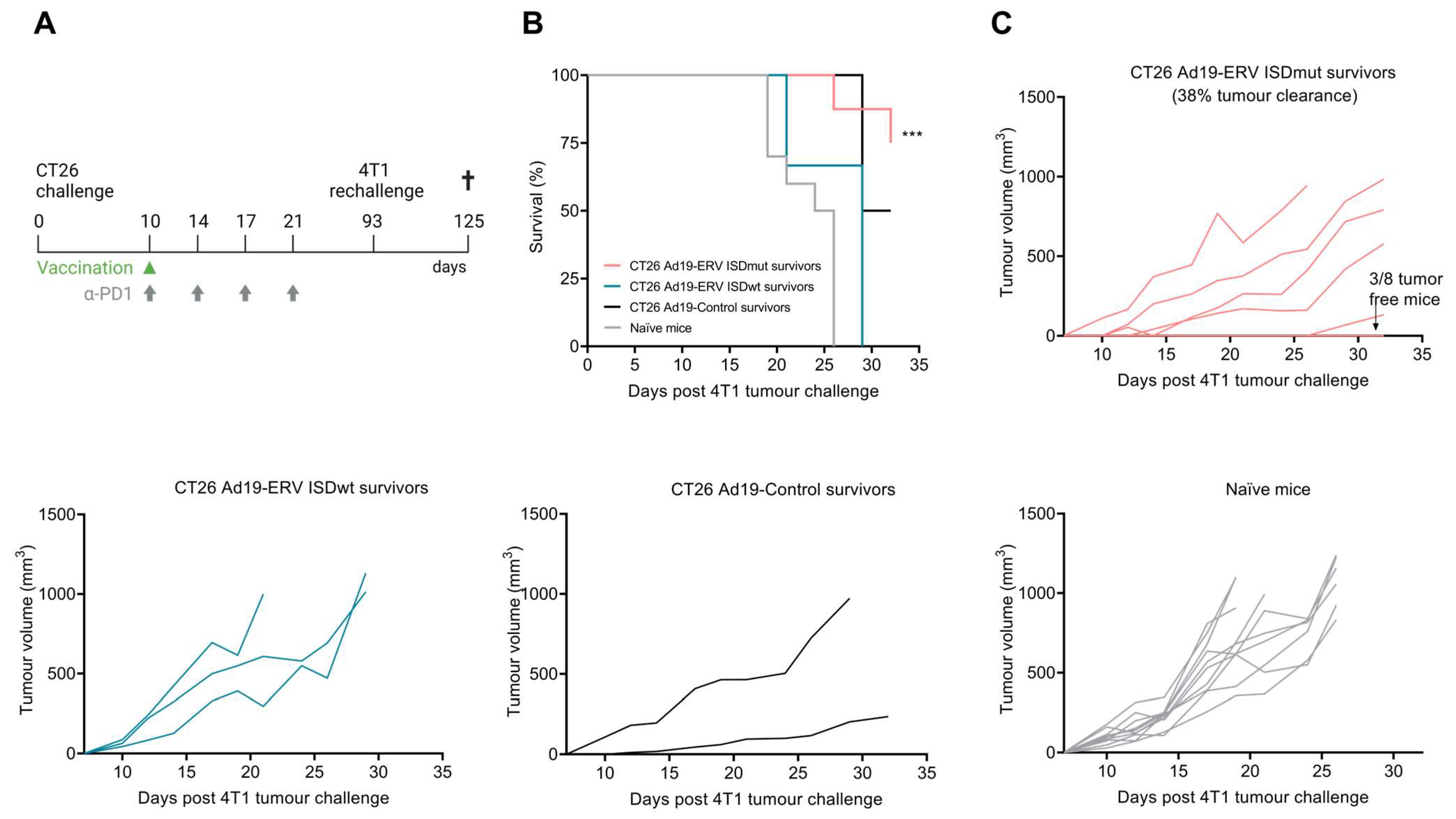
3.5. ISDmut Vaccine Induced Cross-Protection against Different Cancer Types
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-PD1 | anti-PD1 |
| Ad5 | adenovirus type 5; |
| Ad19 or Ad19a or Ad19a/64 | adenovirus type 19a/64; |
| Adv | adenovirus; |
| BMDCs | bone marrow-derived dendritic cells; |
| BSA | bovine serum albumin; |
| CMV | cytomegalovirus promoter; |
| CTL | cytotoxic T lymphocyte; |
| CpG | cytosine-phosphate-guanine; |
| CPI | checkpoint inhibitor; |
| DC | dendritic cell; |
| ELISA | enzyme linked immunosorbent assay; |
| Env | envelope; |
| ERV | endogenous retrovirus; |
| FBS | foetal bovine serum; |
| Gag | group-specific antigen; |
| GM-CSF | granulocyte-macrophage colony stimulating factor; |
| HERV | human endogenous retrovirus; |
| HRP | horseradish peroxidase; |
| IFNγ | interferon gamma; |
| IFU | infectious units; |
| Ig | immunoglobulins; |
| IL | interleukin; |
| i.p. | intraperitoneal; |
| ICS | intracellular cytokine staining; |
| ISD | immunosuppressive domain; |
| ISDmut | immunosuppressive domain mutated; |
| ISDwt | immunosuppressive domain wild type; |
| LPS | lipopolysaccharide; |
| MCS | multiple cloning site; |
| MelARV | melanoma-associated retrovirus; |
| MOI | multiplicity of infection; |
| MuLV | murine leukaemia virus; |
| NaN3 | sodium azide; |
| P2A | porcine teschovirus-1 2A peptide |
| PBS | phosphate buffered saline; |
| s.c. | subcutaneously; |
| SU | surface; |
| SV40-pA | simian virus 40 polyA; |
| TEM | transmission electron microscopy; |
| TNFα | tumour necrosis factor alpha; |
| TM | transmembrane or p15E; |
| UMI | unique molecular identifier |
| VLP | virus-like particle; |
| VLV | virus-like vaccine; |
| WB | Western blot; |
| wt | wild type. |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouse Genome Sequencing Consortium; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocking, C.; Kozak, C.A. Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellåker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Dewannieux, M.; Blaise, S.; Heidmann, T. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J. Virol. 2005, 79, 15573–15577. [Google Scholar] [CrossRef] [Green Version]
- Boller, K.; Schönfeld, K.; Lischer, S.; Fischer, N.; Hoffmann, A.; Kurth, R.; Tönjes, R.R. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J. Gen. Virol. 2008, 89, 567–572. [Google Scholar] [CrossRef]
- Turner, G.; Barbulescu, M.; Su, M.; Jensen-Seaman, M.I.; Kidd, K.K.; Lenz, J. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 2001, 11, 1531–1535. [Google Scholar] [CrossRef] [Green Version]
- Jern, P.; Sperber, G.O.; Blomberg, J. Use of endogenous retroviral sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology 2005, 2, 50. [Google Scholar] [CrossRef] [Green Version]
- Stoye, J.P. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 2012, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Matteucci, C.; Cipriani, C.; Grelli, S.; Ricceri, L.; Calamandrei, G.; Vallebona, P.S. Endogenous Retroviruses Activity as a Molecular Signature of Neurodevelopmental Disorders. Int. J. Mol. Sci. 2019, 20, 6050. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, M.; Gunn, B.M.; Venkataraman, A.; Kong, Y.; Kang, I.; Rakib, T.; Townsend, M.J.; Costenbader, K.H.; Alter, G.; Iwasaki, A. Antibodies against human endogenous retrovirus K102 envelope activate neutrophils in systemic lupus erythematosus. J. Exp. Med. 2021, 218, e20191766. [Google Scholar] [CrossRef] [PubMed]
- Noli, M.; Meloni, G.; Ruberto, S.; Jasemi, S.; Simula, E.R.; Cossu, D.; Bo, M.; Palermo, M.; Sechi, L.A. HERV-K Envelope Protein Induces Long-Lasting Production of Autoantibodies in T1DM Patients at Onset in Comparison to ZNT8 Autoantibodies. Pathogens 2022, 11, 1188. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Erbì, M.C.; Scognamiglio, S.; Tramontano, E. Human Endogenous Retrovirus (HERV) Transcriptome Is Dynamically Modulated during SARS-CoV-2 Infection and Allows Discrimination of COVID-19 Clinical Stages. Microbiol. Spectr. 2023, 11, e0251622. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.D.; Holst, P.J.; Nielsen, K.N.; Dons Müller, M.; Holst, P.J.; Nielsen, K.N.; Müller, M.D.; Holst, P.J.; Nielsen, K.N. A Systematic Review of Expression and Immunogenicity of Human Endogenous Retroviral Proteins in Cancer and Discussion of Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 1330. [Google Scholar] [CrossRef]
- Kassiotis, G. Endogenous retroviruses and the development of cancer. J. Immunol. 2014, 192, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Pothlichet, J.; Mangeney, M.; Heidmann, T. Mobility and integration sites of a murine C57BL/6 melanoma endogenous retrovirus involved in tumor progression in vivo. Int. J. Cancer 2006, 119, 1869–1877. [Google Scholar] [CrossRef]
- Scrimieri, F.; Askew, D.; Corn, D.J.; Eid, S.; Bobanga, I.D.; Bjelac, J.A.; Tsao, M.L.; Allen, F.; Othman, Y.S.; Wang, S.-C.G.; et al. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. Oncoimmunology 2013, 2, e26889. [Google Scholar] [CrossRef]
- Huang, A.Y.; Gulden, P.H.; Woods, A.S.; Thomas, M.C.; Tong, C.D.; Wang, W.; Engelhard, V.H.; Pasternack, G.; Cotter, R.; Hunt, D.; et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc. Natl. Acad. Sci. USA 1996, 93, 9730–9735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Huang, X.; Zhu, Z.; Gorelik, E. Sequence and insertion sites of murine melanoma-associated retrovirus. J. Virol. 1999, 73, 9178–9186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecht-Louf, G.; Renard, M.; Mangeney, M.; Letzelter, C.; Richaud, A.; Ducos, B.; Bouallaga, I.; Heidmann, T. Retroviral infection in vivo requires an immune escape virulence factor encrypted in the envelope protein of oncoretroviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 3782–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morozov, V.A.; Morozov, A.V.; Semaan, M.; Denner, J. Single mutations in the transmembrane envelope protein abrogate the immunosuppressive property of HIV-1. Retrovirology 2012, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangeney, M.; Heidmann, T. Tumor cells expressing a retroviral envelope escape immune rejection in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 14920–14925. [Google Scholar] [CrossRef] [Green Version]
- Mangeney, M.; Renard, M.; Schlecht-Louf, G.; Bouallaga, I.; Heidmann, O.; Letzelter, C.; Richaud, A.; Ducos, B.; Heidmann, T. Placental syncytins: Genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20534–20539. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.S.; Hovenkamp, E.; Savelkoul, H.F.; Knegt, P.; Van Ewijk, W. Immunotherapy with monoclonal antibodies directed against the immunosuppressive domain of p15E inhibits tumour growth. Clin. Exp. Immunol. 1995, 102, 468–475. [Google Scholar] [CrossRef]
- Eisenthal, A.; Lafreniere, R.; Lefor, A.T.; Rosenberg, S.A. Effect of anti-B16 melanoma monoclonal antibody on established murine B16 melanoma liver metastases. Cancer Res. 1987, 47, 2771–2776. [Google Scholar] [PubMed]
- Hearing, V.J.; Leong, S.P.; Vieira, W.D.; Law, L.W. Suppression of established pulmonary metastases by murine melanoma-specific monoclonal antibodies. Int. J. Cancer 1991, 47, 148–153. [Google Scholar] [CrossRef]
- Marcus, S.G.; Perry-Lalley, D.; Mulé, J.J.; Rosenberg, S.A.; Yang, J.C. The use of interleukin-6 to generate tumor-infiltrating lymphocytes with enhanced in vivo antitumor activity. J. Immunother. Emphas. Tumor Immunol. 1994, 15, 105–112. [Google Scholar] [CrossRef]
- Yang, S.; Vervaert, C.E.; Burch, J.; Grichnik, J.; Seigler, H.F.; Darrow, T.L. Murine dendritic cells transfected with human GP100 elicit both antigen- specific CD8+ and CD4+ T-cell responses and are more effective than DNA vaccines at generating anti-tumor immunity. Int. J. Cancer 1999, 83, 532–540. [Google Scholar] [CrossRef]
- Yang, J.C.; Perry-Lalley, D. The envelope protein of an endogenous murine retrovirus is a tumor-associated T-cell antigen for multiple murine tumors. J. Immunother. 2000, 23, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Cingarlini, S.; Apolloni, E.; Serafini, P.; Marigo, I.; De Santo, C.; Macino, B.; Marin, O.; Zanovello, P.; De Santo, C.; et al. Effective genetic vaccination with a widely shared endogenous retroviral tumor antigen requires CD40 stimulation during tumor rejection phase. J. Immunol. 2003, 171, 6396–6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slansky, J.E.; Rattis, F.M.; Boyd, L.F.; Fahmy, T.; Jaffee, E.M.; Schneck, J.P.; Margulies, D.H.; Pardoll, D.M. Enhanced Antigen-Specific Antitumor Immunity with Altered Peptide Ligands that Stabilize the MHC-Peptide-TCR Complex. Immunity 2000, 13, 529–538. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Zhou, S.; Quinn, B.; Huang, W.-C.C.; Jahagirdar, D.; Vega, M.; Ortega, J.; Long, M.D.; Ito, F.; Abrams, S.I.; et al. Position-Scanning Peptide Libraries as Particle Immunogens for Improving CD8+ T-Cell Responses. Adv. Sci. 2021, 8, e2103023. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Mondesire, W.; Wang, G.; Overwijk, W.W.; Lapointe, R.; Yang, J.C.; Restifo, N.P.; Hwu, P.; Hsu, C.; Parker, L.L.; et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res. 2001, 61, 7920–7924. [Google Scholar]
- Thiel, H.J.; Schwarz, H.; Fischinger, P.; Bolognesi, D.; Schäfer, W. Role of antibodies to murine leukemia virus p15E transmembrane protein in immunotherapy against AKR leukemia: A model for studies in human acquired immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1987, 84, 5893–5897. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.-M.C.; Holst, P.J. Increased T cell breadth and antibody response elicited in prime-boost regimen by viral vector encoded homologous SIV Gag/Env in outbred CD1 mice. J. Transl. Med. 2016, 14, 343. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.-M.C.; Resende, M.; Salanti, A.; Nielsen, M.A.; Holst, P.J. Novel adenovirus encoded virus-like particles displaying the placental malaria associated VAR2CSA antigen. Vaccine 2017, 35, 1140–1147. [Google Scholar] [CrossRef]
- Andersson, A.-M.C.; Schwerdtfeger, M.; Holst, P.J. Virus-Like-Vaccines against HIV. Vaccines 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Neukirch, L.; Nielsen, T.K.; Laursen, H.; Daradoumis, J.; Thirion, C.; Holst, P.J. Adenovirus based virus-like-vaccines targeting endogenous retroviruses can eliminate growing colorectal cancers in mice. Oncotarget 2019, 10, 1458–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzsics, Z.; Lemnitzer, F.; Thirion, C. Engineering adenovirus genome by bacterial artificial chromosome (BAC) technology. Methods Mol Biol. 2014, 1089, 143–158. [Google Scholar] [CrossRef] [PubMed]
- D’Alise, A.M.; Leoni, G.; Cotugno, G.; Troise, F.; Langone, F.; Fichera, I.; De Lucia, M.; Avalle, L.; Vitale, R.; Leuzzi, A.; et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun. 2019, 10, 2688. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Sprent, J. GM-CSF Culture Revisited: Preparation of Bulk Populations of Highly Pure Dendritic Cells from Mouse Bone Marrow. J. Immunol. 2018, 201, 3129–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Janik, P.; Briand, P.; Hartmann, N.R. The Effect of Estrone-Progesterone Treatment on Cell Proliferation Kinetics of Hormone-dependent GR Mouse Mammary Tumors. Cancer Res. 1975, 35, 3698–3704. [Google Scholar]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef]
- Zhou, F.; Li, M.; Wei, Y.; Lin, K.; Lu, Y.; Shen, J.; Johanning, G.L.; Wang-Johanning, F. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget 2016, 7, 84093–84117. [Google Scholar] [CrossRef] [Green Version]
- Wang-Johanning, F.; Frost, A.R.; Jian, B.; Epp, L.; Lu, D.W.; Johanning, G.L. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene 2003, 22, 1528–1535. [Google Scholar] [CrossRef] [Green Version]
- Wang-Johanning, F.; Liu, J.; Rycaj, K.; Huang, M.; Tsai, K.; Rosen, D.G.; Chen, D.-T.; Lu, D.W.; Barnhart, K.F.; Johanning, G.L. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int. J. Cancer 2007, 120, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Manca, M.A.; Solinas, T.; Simula, E.R.; Noli, M.; Ruberto, S.; Madonia, M.; Sechi, L.A. HERV-K and HERV-H Env Proteins Induce a Humoral Response in Prostate Cancer Patients. Pathogens 2022, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Winnenthal, F.H.; Galindo-Escobedo, L.V.; Rimoldi, D.; Geng, W.; Romero, P.; Koch, M.; Weitz, J.; Krempien, R.; Niethammer, A.G.; Beckhove, P.; et al. Potential target antigens for immunotherapy in human pancreatic cancer. Cancer Lett. 2007, 252, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Serafino, A.; Balestrieri, E.; Pierimarchi, P.; Matteucci, C.; Moroni, G.; Oricchio, E.; Rasi, G.; Mastino, A.; Spadafora, C.; Garaci, E.; et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp. Cell Res. 2009, 315, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Downey, R.F.; Sullivan, F.J.; Wang-Johanning, F.; Ambs, S.; Giles, F.J.; Glynn, S.A. Human endogenous retrovirus K and cancer: Innocent bystander or tumorigenic accomplice? Int. J. Cancer 2015, 137, 1249–1257. [Google Scholar] [CrossRef] [Green Version]
- Rhyu, D.-W.; Kang, Y.-J.; Ock, M.-S.; Eo, J.-W.; Choi, Y.-H.; Kim, W.-J.; Leem, S.-H.; Yi, J.-M.; Kim, H.-S.; Cha, H.-J. Expression of human endogenous retrovirus env genes in the blood of breast cancer patients. Int. J. Mol. Sci. 2014, 15, 9173–9183. [Google Scholar] [CrossRef] [Green Version]
- Menendez, L.; Benigno, B.B.; McDonald, J.F. L1 and HERV-W retrotransposons are hypomethylated in human ovarian carcinomas. Mol. Cancer 2004, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, J.; Montgiraud, C.; Pichon, J.-P.; Bonnaud, B.; Arsac, M.; Ruel, K.; Bouton, O.; Mallet, F. Custom human endogenous retroviruses dedicated microarray identifies self-induced HERV-W family elements reactivated in testicular cancer upon methylation control. Nucleic Acids Res. 2010, 38, 2229–2246. [Google Scholar] [CrossRef]
- Pérot, P.; Mullins, C.S.; Naville, M.; Bressan, C.; Hühns, M.; Gock, M.; Kühn, F.; Volff, J.-N.; Trillet-Lenoir, V.; Linnebacher, M.; et al. Expression of young HERV-H loci in the course of colorectal carcinoma and correlation with molecular subtypes. Oncotarget 2015, 6, 40095–40111. [Google Scholar] [CrossRef] [Green Version]
- Mangeney, M.; de Parseval, N.; Thomas, G.; Heidmann, T. The full-length envelope of an HERV-H human endogenous retrovirus has immunosuppressive properties. J. Gen. Virol. 2001, 82, 2515–2518. [Google Scholar] [CrossRef] [Green Version]
- Eksmond, U.; Jenkins, B.; Merkenschlager, J.; Mothes, W.; Stoye, J.P.; Kassiotis, G. Mutation of the Putative Immunosuppressive Domain of the Retroviral Envelope Glycoprotein Compromises Infectivity. J. Virol. 2017, 91, e01152-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Hogan, V.; Johnson, W.E. Unique Structure and Distinctive Properties of the Ancient and Ubiquitous Gamma-Type Envelope Glycoprotein. Viruses 2023, 15, 274. [Google Scholar] [CrossRef]
- Chirmule, N.; Propert, K.; Magosin, S.; Qian, Y.; Qian, R.; Wilson, J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999, 6, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Ng’ang’a, D.; Brandariz, K.L.; Abbink, P.; et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef]
- Sato, Y.; Fu, Y.; Liu, H.; Lee, M.Y.; Shaw, M.H. Tumor-immune profiling of CT-26 and Colon 26 syngeneic mouse models reveals mechanism of anti-PD-1 response. BMC Cancer 2021, 21, 1222. [Google Scholar] [CrossRef]
- Maine, C.J.; Richard, G.; Spasova, D.S.; Miyake-Stoner, S.J.; Sparks, J.; Moise, L.; Sullivan, R.P.; Garijo, O.; Choz, M.; Crouse, J.M.; et al. Self-Replicating RNAs Drive Protective Anti-tumor T Cell Responses to Neoantigen Vaccine Targets in a Combinatorial Approach. Mol. Ther. 2021, 29, 1186–1198. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Baklaushev, V.P.; Kilpeläinen, A.; Petkov, S.; Abakumov, M.A.; Grinenko, N.F.; Yusubalieva, G.M.; Latanova, A.A.; Gubskiy, I.L.; Zabozlaev, F.G.; Starodubova, E.S.; et al. Luciferase Expression Allows Bioluminescence Imaging But Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci. Rep. 2017, 7, 7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottina, E.; Levy, P.; Eksmond, U.; Merkenschlager, J.; Young, G.R.; Roels, J.; Stoye, J.P.; Tüting, T.; Calado, D.P.; Kassiotis, G. Restoration of Endogenous Retrovirus Infectivity Impacts Mouse Cancer Models. Cancer Immunol. Res. 2018, 6, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Alcazer, V.; Mutez, V.; Tonon, L.; Martin, J.; Chuvin, N.; Michel, E.; Boulos, R.E.; Estornes, Y.; Valladeau-Guilemond, J.; et al. Identification of shared tumor epitopes from endogenous retroviruses inducing high-avidity cytotoxic T cells for cancer immunotherapy. Sci. Adv. 2022, 8, eabj3671. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daradoumis, J.; Ragonnaud, E.; Skandorff, I.; Nielsen, K.N.; Bermejo, A.V.; Andersson, A.-M.; Schroedel, S.; Thirion, C.; Neukirch, L.; Holst, P.J. An Endogenous Retrovirus Vaccine Encoding an Envelope with a Mutated Immunosuppressive Domain in Combination with Anti-PD1 Treatment Eradicates Established Tumours in Mice. Viruses 2023, 15, 926. https://doi.org/10.3390/v15040926
Daradoumis J, Ragonnaud E, Skandorff I, Nielsen KN, Bermejo AV, Andersson A-M, Schroedel S, Thirion C, Neukirch L, Holst PJ. An Endogenous Retrovirus Vaccine Encoding an Envelope with a Mutated Immunosuppressive Domain in Combination with Anti-PD1 Treatment Eradicates Established Tumours in Mice. Viruses. 2023; 15(4):926. https://doi.org/10.3390/v15040926
Chicago/Turabian StyleDaradoumis, Joana, Emeline Ragonnaud, Isabella Skandorff, Karen Nørgaard Nielsen, Amaia Vergara Bermejo, Anne-Marie Andersson, Silke Schroedel, Christian Thirion, Lasse Neukirch, and Peter Johannes Holst. 2023. "An Endogenous Retrovirus Vaccine Encoding an Envelope with a Mutated Immunosuppressive Domain in Combination with Anti-PD1 Treatment Eradicates Established Tumours in Mice" Viruses 15, no. 4: 926. https://doi.org/10.3390/v15040926