
HIV-1 Group M Capsid Amino Acid Variability: Implications for Sequence Quality Control of Genotypic Resistance Testing

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Creating a Consensus Sequence
2.3. Sequence Quality Control
2.4. Inter- and Intra-Subtype Variation
2.5. Amino Acid Profile
2.6. Correlation Analysis
3. Results
3.1. References, Patients, and Sequences
3.2. Sequence Quality Control
3.3. Inter- and Intra-Subtype Variability
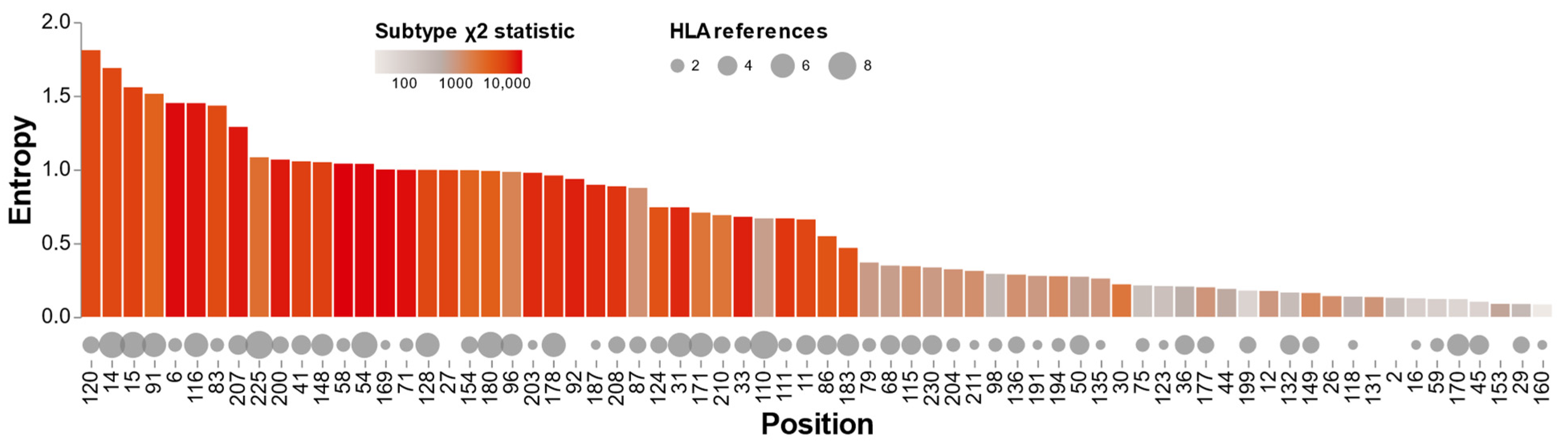
3.4. Capsid Amino Acid Profile
3.5. HLA-Associated Polymorphisms and Subtype Variability
3.6. Correlated Positions
3.7. Database and Sequence Interpretation Program
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef]
- McFadden, W.M.; Snyder, A.A.; Kirby, K.A.; Tedbury, P.R.; Raj, M.; Wang, Z.; Sarafianos, S.G. Rotten to the core: Antivirals targeting the HIV-1 capsid core. Retrovirology 2021, 18, 41. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yamashita, M. HIV-1 capsid variability: Viral exploitation and evasion of capsid-binding molecules. Retrovirology 2021, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2022, 386, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Struck, D.; Lawyer, G.; Ternes, A.-M.; Schmit, J.-C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic. Acids. Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.; Vanderveen, L.; Naik, V.; Ram, R.; Parvangada, P.; Martin, R.; Rhee, M.; Callebaut, C. Phenotypic resistance to lenacapavir and monotherapy efficacy in a proof-of-concept clinical study. J. Antimicrob. Chemother. 2022, 77, 989–995. [Google Scholar] [CrossRef]
- Rose, P.P.; Korber, B.T. Detecting hypermutations in viral sequences with an emphasis on G --> A hypermutation. Bioinformatics 2000, 16, 400–401. [Google Scholar] [CrossRef]
- Rhee, S.-Y.; Sankaran, K.; Varghese, V.; Winters, M.A.; Hurt, C.B.; Eron, J.J.; Parkin, N.; Holmes, S.P.; Holodniy, M.; Shafer, R.W. HIV-1 Protease, Reverse Transcriptase, and Integrase Variation. J. Virol. 2016, 90, 6058–6070. [Google Scholar] [CrossRef]
- Brumme, Z.L.; John, M.; Carlson, J.M.; Brumme, C.J.; Chan, D.; Brockman, M.A.; Swenson, L.C.; Tao, I.; Szeto, S.; Rosato, P.; et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS ONE 2009, 4, e6687. [Google Scholar] [CrossRef]
- Chikata, T.; Carlson, J.M.; Tamura, Y.; Borghan, M.A.; Naruto, T.; Hashimoto, M.; Murakoshi, H.; Le, A.Q.; Mallal, S.; John, M.; et al. Host-Specific Adaptation of HIV-1 Subtype B in the Japanese Population. J. Virol. 2014, 88, 4764–4775. [Google Scholar] [CrossRef]
- Kinloch, N.N.; Lee, G.Q.; Carlson, J.M.; Jin, S.W.; Brumme, C.J.; Byakwaga, H.; Muzoora, C.; Bwana, M.B.; Cobarrubias, K.D.; Hunt, P.W.; et al. Genotypic and Mechanistic Characterization of Subtype-Specific HIV Adaptation to Host Cellular Immunity. J. Virol. 2018, 93, e01502-18. [Google Scholar] [CrossRef]
- Rousseau, C.M.; Daniels, M.G.; Carlson, J.M.; Kadie, C.; Crawford, H.; Prendergast, A.; Matthews, P.; Payne, R.; Rolland, M.; Raugi, D.N.; et al. HLA Class I-Driven Evolution of Human Immunodeficiency Virus Type 1 Subtype C Proteome: Immune Escape and Viral Load. J. Virol. 2008, 82, 6434–6446. [Google Scholar] [CrossRef]
- Van Tran, G.; Chikata, T.; Carlson, J.M.; Murakoshi, H.; Nguyen, D.H.; Tamura, Y.; Akahoshi, T.; Kuse, N.; Sakai, K.; Sakai, S.; et al. A strong association of human leukocyte antigen-associated Pol and Gag mutations with clinical parameters in HIV-1 subtype A/E infection. AIDS 2016, 30, 681–689. [Google Scholar] [CrossRef]
- Gesprasert, G.; Wichukchinda, N.; Mori, M.; Shiino, T.; Auwanit, W.; Sriwanthana, B.; Pathipvanich, P.; Sawanpanyalert, P.; Miura, T.; Auewarakul, P.; et al. HLA-Associated Immune Pressure on Gag Protein in CRF01_AE-Infected Individuals and Its Association with Plasma Viral Load. PLoS ONE 2010, 5, e11179. [Google Scholar] [CrossRef]
- Carlson, J.M.; Brumme, Z.L.; Rousseau, C.M.; Brumme, C.J.; Matthews, P.; Kadie, C.; Mullins, J.I.; Walker, B.D.; Harrigan, P.R.; Goulder, P.J.R.; et al. Phylogenetic Dependency Networks: Inferring Patterns of CTL Escape and Codon Covariation in HIV-1 Gag. PLoS Comput. Biol. 2008, 4, e1000225. [Google Scholar] [CrossRef]
- Carlson, J.M.; Schaefer, M.; Monaco, D.C.; Batorsky, R.; Claiborne, D.T.; Prince, J.; Deymier, M.J.; Ende, Z.S.; Klatt, N.R.; DeZiel, C.E.; et al. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 2014, 345, 1254031. [Google Scholar] [CrossRef]
- Carlson, J.M.; Listgarten, J.; Pfeifer, N.; Tan, V.; Kadie, C.; Walker, B.D.; Ndung’u, T.; Shapiro, R.; Frater, J.; Brumme, Z.L.; et al. Widespread Impact of HLA Restriction on Immune Control and Escape Pathways of HIV-1. J. Virol. 2012, 86, 5230–5243. [Google Scholar] [CrossRef]
- Carlson, J.M.; Le, A.Q.; Shahid, A.; Brumme, Z.L. HIV-1 adaptation to HLA: A window into virus–host immune interactions. Trends. Microbiol. 2015, 23, 212–224. [Google Scholar] [CrossRef]
- Kloke, J.D.; McKean, J.W. Rfit: Rank-based Estimation for Linear Models. R J. 2012, 4, 57. [Google Scholar] [CrossRef]
- Poon, A.F.Y.; Lewis, F.I.; Frost, S.D.W.; Kosakovsky Pond, S.L. Spidermonkey: Rapid detection of co-evolving sites using Bayesian graphical models. Bioinformatics 2008, 24, 1949–1950. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-Ray Structures of the Hexameric Building Block of the HIV Capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef]
- Wagner, J.M.; Zadrozny, K.K.; Chrustowicz, J.; Purdy, M.D.; Yeager, M.; Ganser-Pornillos, B.K.; Pornillos, O. Crystal structure of an HIV assembly and maturation switch. eLife 2016, 5, e17063. [Google Scholar] [CrossRef]
- Schur, F.K.M.; Obr, M.; Hagen, W.J.H.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Kräusslich, H.-G.; Briggs, J.A.G. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef]
- Bester, S.M.; Adu-Ampratwum, D.; Annamalai, A.S.; Wei, G.; Briganti, L.; Murphy, B.C.; Haney, R.; Fuchs, J.R.; Kvaratskhelia, M. Structural and Mechanistic Bases of Viral Resistance to HIV-1 Capsid Inhibitor Lenacapavir. mBio 2022, 13, e01804-22. [Google Scholar] [CrossRef]
- Perilla, J.R.; Gronenborn, A.M. Molecular Architecture of the Retroviral Capsid. Trends. Biochem. Sci. 2016, 41, 410–420. [Google Scholar] [CrossRef]
- Leslie, A.J.; Pfafferott, K.J.; Chetty, P.; Draenert, R.; Addo, M.M.; Feeney, M.; Tang, Y.; Holmes, E.C.; Allen, T.; Prado, J.G.; et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 2004, 10, 282–289. [Google Scholar] [CrossRef]
- Tzou, P.L.; Pond, S.L.K.; Avila-Rios, S.; Holmes, S.P.; Kantor, R.; Shafer, R.W. Analysis of unusual and signature APOBEC-mutations in HIV-1 pol next-generation sequences. PLoS ONE 2020, 15, e0225352. [Google Scholar] [CrossRef]
- Rihn, S.J.; Wilson, S.J.; Loman, N.J.; Alim, M.; Bakker, S.E.; Bhella, D.; Gifford, R.J.; Rixon, F.J.; Bieniasz, P.D. Extreme Genetic Fragility of the HIV-1 Capsid. PLOS Pathog. 2013, 9, e1003461. [Google Scholar] [CrossRef]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; de Pierres, C.; Mncube, Z.; Mkhwanazi, N.; et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef]
- International HIV Controllers Study; Pereyra, F.; Jia, X.; McLaren, P.J.; Telenti, A.; de Bakker, P.I.W.; Walker, B.D.; Ripke, S.; Brumme, C.J.; Pulit, S.L.; et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 2010, 330, 1551–1557. [Google Scholar] [CrossRef]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Fleminger, I.; Kirtley, S.; Williams, B.; Gouws-Williams, E.; Ghys, P.D.; Abimiku, A.G.; et al. Global and regional molecular epidemiology of HIV-1, 1990–2015: A systematic review, global survey, and trend analysis. Lancet. Infect. Dis. 2019, 19, 143–155. [Google Scholar] [CrossRef]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360–364. [Google Scholar] [CrossRef]
- Gupta, S.K.; Berhe, M.; Crofoot, G.; Benson, P.; Ramgopal, M.; Sims, J.; McDonald, C.; Ruane, P.; Sanchez, W.E.; Scribner, A.; et al. Lenacapavir administered every 26 weeks or daily in combination with oral daily antiretroviral therapy for initial treatment of HIV: A randomised, open-label, active-controlled, phase 2 trial. Lancet. HIV 2023, 10, e15–e23. [Google Scholar] [CrossRef]
- Li, G.; Verheyen, J.; Rhee, S.-Y.; Voet, A.; Vandamme, A.-M.; Theys, K. Functional conservation of HIV-1 Gag: Implications for rational drug design. Retrovirology 2013, 10, 126. [Google Scholar] [CrossRef]
- Troyano-Hernáez, P.; Reinosa, R.; Holguín, Á. HIV Capsid Protein Genetic Diversity Across HIV-1 Variants and Impact on New Capsid-Inhibitor Lenacapavir. Front. Microbiol. 2022, 13, 854974. [Google Scholar] [CrossRef]
- Marcelin, A.-G.; Charpentier, C.; Jary, A.; Perrier, M.; Margot, N.; Callebaut, C.; Calvez, V.; Descamps, D. Frequency of capsid substitutions associated with GS-6207 in vitro resistance in HIV-1 from antiretroviral-naive and -experienced patients. J. Antimicrob. Chemother. 2020, 75, 1588–1590. [Google Scholar] [CrossRef]
- Nka, A.D.; Bouba, Y.; Teto, G.; Semengue, E.N.J.; Takou, D.K.; Ngueko, A.M.K.; Fabeni, L.; Carioti, L.; Armenia, D.; Pabo, W.; et al. Evaluation of HIV-1 capsid genetic variability and lenacapavir (GS-6207) drug resistance-associated mutations according to viral clades among drug-naive individuals. J. Antimicrob. Chemother. 2022, 78, 272–275. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Sequences (n = 21,012) |
|---|---|
| Subtype | n (%) |
| A | 3841 (18) |
| B | 5716 (27) |
| C | 4806 (23) |
| D | 1668 (8.0) |
| F | 399 (1.9) |
| G | 291 (1.4) |
| CRF01_AE | 2967 (14) |
| CRF02_AG | 84 (0.4) |
| Other CRF | 956 (4.6) |
| URF | 249 (1.2) |
| H | 21 (0.10) |
| J | 6 (0.03) |
| K | 7 (0.03) |
| L | 1 (0.005) |
| Treatment histories | |
| None | 19,199 (91.4) |
| RTI | 442 (2.1) |
| PI | 323 (1.5) |
| Unknown | 1371 (6.5) |
| Sequencing method | |
| PCR dideoxy terminator sequencing | 19,281 (91.8) |
| Single or consensus of molecular clonal sequencing | 1211 (5.8) |
| Single or consensus of single genome sequences | 420 (2.0) |
| Consensus of NGS | 100 (0.5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, K.; Rhee, S.-Y.; Tzou, P.L.; Osman, Z.A.; Pond, S.L.K.; Holmes, S.P.; Shafer, R.W. HIV-1 Group M Capsid Amino Acid Variability: Implications for Sequence Quality Control of Genotypic Resistance Testing. Viruses 2023, 15, 992. https://doi.org/10.3390/v15040992
Tao K, Rhee S-Y, Tzou PL, Osman ZA, Pond SLK, Holmes SP, Shafer RW. HIV-1 Group M Capsid Amino Acid Variability: Implications for Sequence Quality Control of Genotypic Resistance Testing. Viruses. 2023; 15(4):992. https://doi.org/10.3390/v15040992
Chicago/Turabian StyleTao, Kaiming, Soo-Yon Rhee, Philip L. Tzou, Zachary A. Osman, Sergei L. Kosakovsky Pond, Susan P. Holmes, and Robert W. Shafer. 2023. "HIV-1 Group M Capsid Amino Acid Variability: Implications for Sequence Quality Control of Genotypic Resistance Testing" Viruses 15, no. 4: 992. https://doi.org/10.3390/v15040992
APA StyleTao, K., Rhee, S.-Y., Tzou, P. L., Osman, Z. A., Pond, S. L. K., Holmes, S. P., & Shafer, R. W. (2023). HIV-1 Group M Capsid Amino Acid Variability: Implications for Sequence Quality Control of Genotypic Resistance Testing. Viruses, 15(4), 992. https://doi.org/10.3390/v15040992