
Molecular Population Genetics of Aspen Mosaic-Associated Virus in Finland and Sweden

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Source
2.2. Primers, Detection of AsMaV by RT–PCR
2.3. AsMaV–RNA3 RT–PCR–RFLP
2.4. Amplification of Different Genomic Regions
2.5. Cloning, Sequencing, and Data Analysis
3. Results and Discussion
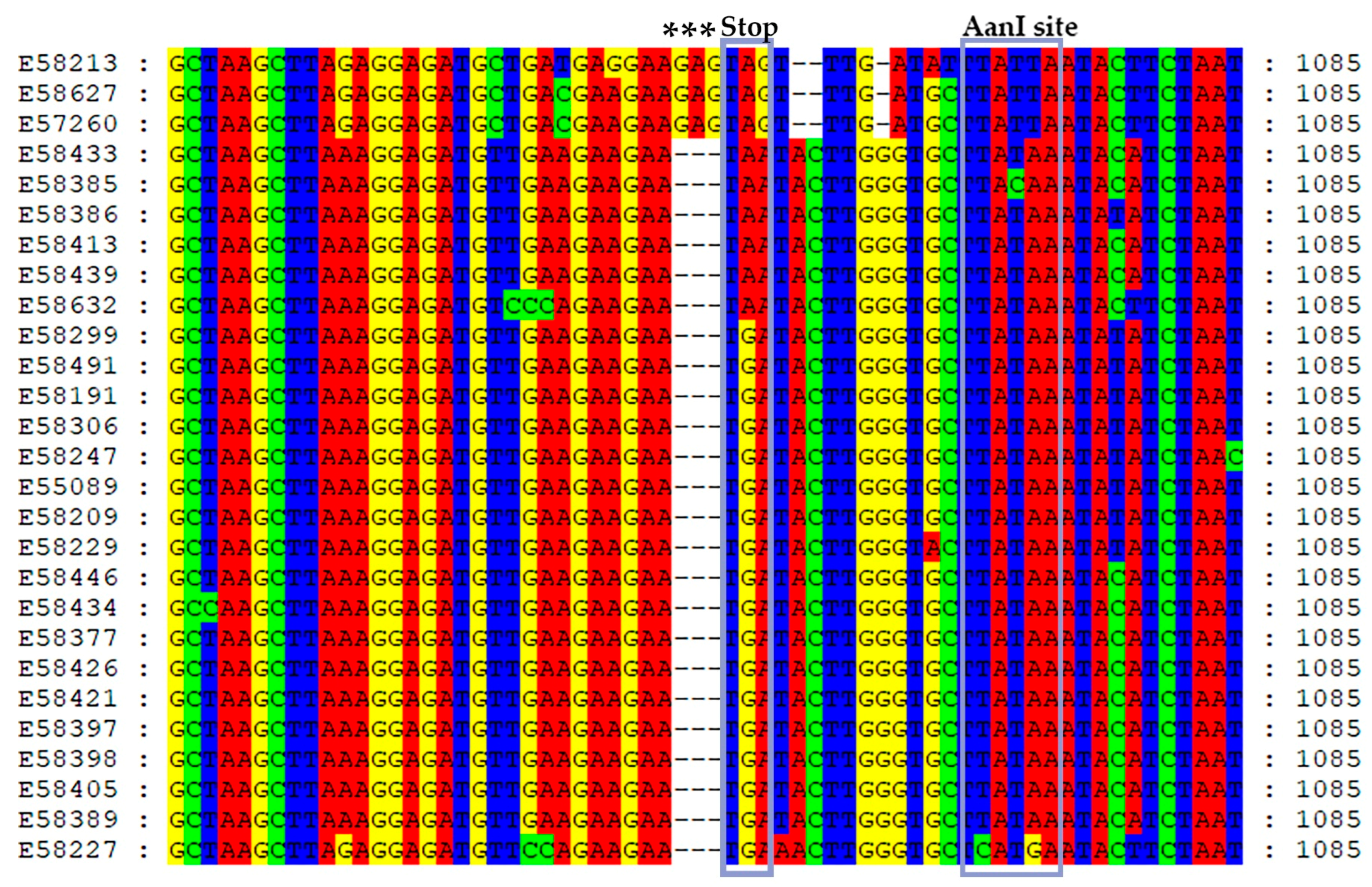
3.1. Detection of AsMaV and RT–PCR–RFLP
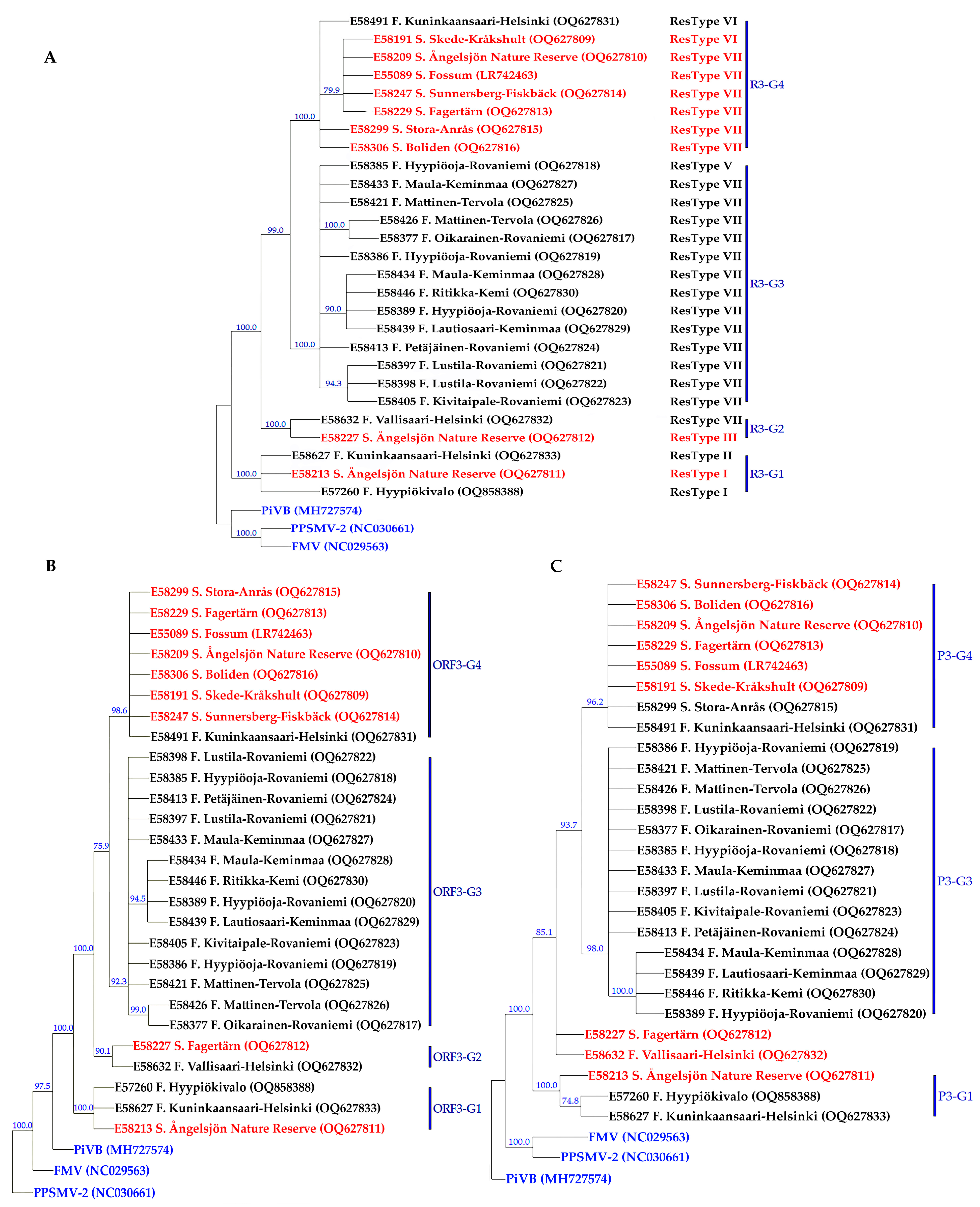
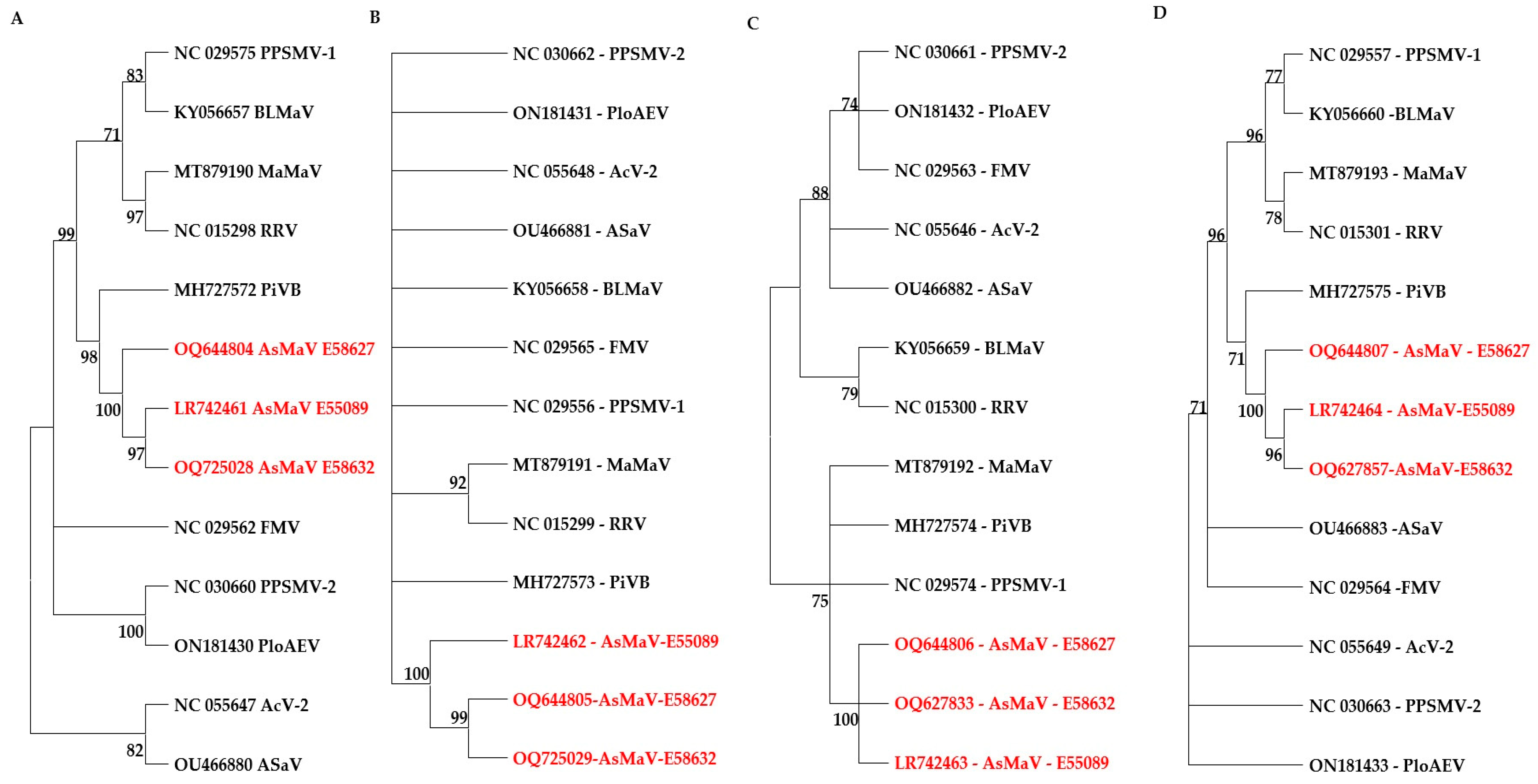
3.2. Genetic Diversity and Phylogeny of AsMaV Isolates Based on RNA3
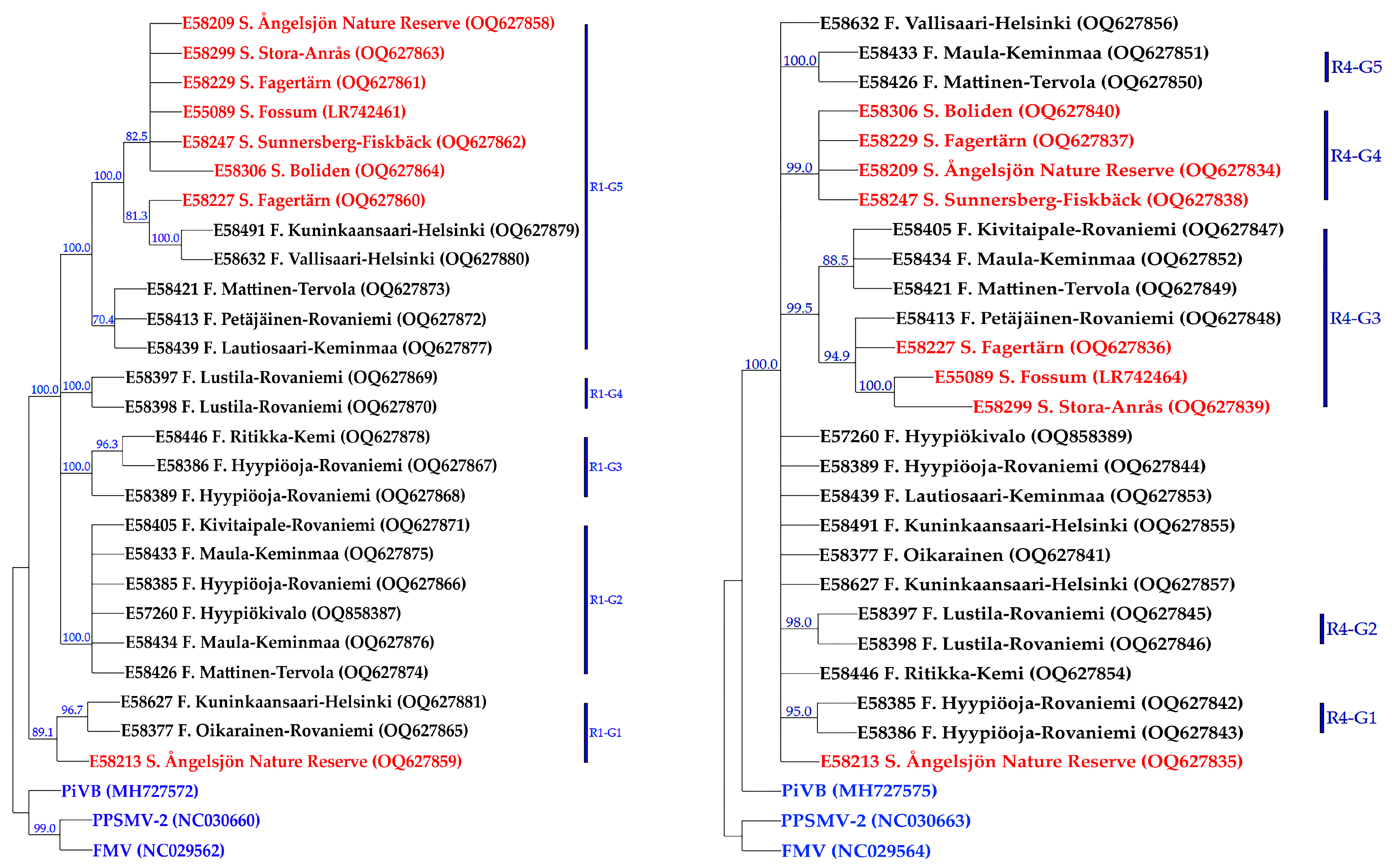
3.3. Genetic Diversity and Phylogeny of AsMaV Isolates Based on RNA4
3.4. Genetic Diversity and Phylogeny of AsMaV Isolates Based on Partial RNA1
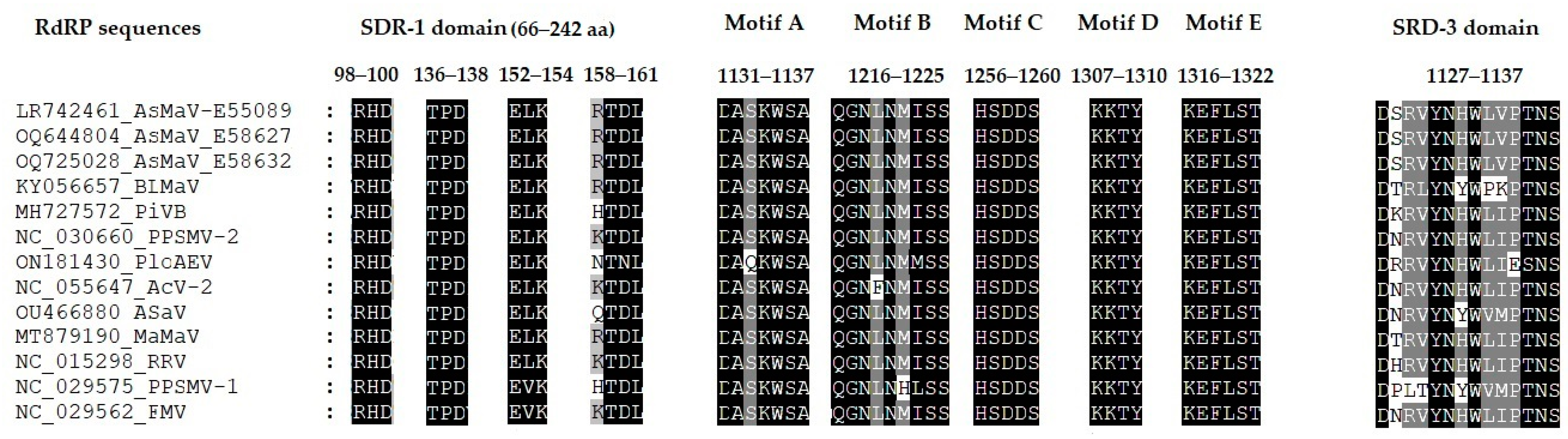
3.5. Complete Sequence of AsMaV-RNA1, RNA2 and RNA 5 of Distant Isolates
3.6. Relation of Distant Isolates of AsMaV with Clade A Emaraviruses
3.7. Population Genetic Parameters
3.8. Population Differentiation Parameters
3.9. Recombination Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caudullo, G.; De Rigo, D. Populus tremula in Europe: Distribution, habitat, usage and threats. In European Atlas of Forest Tree Species; San-Miguel-Ayanz, J., de Rigo, D., Caudullo, G., Houston Durrant, T., Mauri, A., Eds.; Publications Office of the European Union: Luxembourg, 2016. [Google Scholar]
- Rogers, P.C.; Pinno, B.D.; Šebesta, J.; Albrectsen, B.R.; Li, G.; Ivanova, N.; Kusbach, A.; Kuuluvainen, T.; Landhäusser, S.M.; Liu, H.; et al. A global view of aspen: Conservation science for widespread keystone systems. Glob. Ecol. Conserv. 2020, 21, e00828. [Google Scholar] [CrossRef]
- Kivinen, S.; Koivisto, E.; Keski-Saari, S.; Poikolainen, L.; Tanhuanpää, T.; Kuzmin, A.; Viinikka, A.; Heikkinen, R.K.; Pykälä, J.; Virkkala, R.; et al. A keystone species, European aspen (Populus tremula L.), in boreal forests: Ecological role, knowledge needs and mapping using remote sensing. For. Ecol. Manag. 2020, 462, 118008. [Google Scholar] [CrossRef]
- Jactel, H.; Koricheva, J.; Castagneyrol, B. Responses of forest insect pests to climate change: Not so simple. Curr. Opin. Insect Sci. 2019, 35, 103–108. [Google Scholar] [CrossRef]
- von Bargen, S.; Al Kubrusli, R.; Gaskin, T.; Fürl, S.; Hüttner, F.; Blystad, D.R.; Karlin, D.G.; Jalkanen, R.; Büttner, C. Characterisation of a novel Emaravirus identified in mosaic-diseased Eurasian aspen (Populus tremula). Ann. Appl. Biol. 2020, 176, 210–222. [Google Scholar] [CrossRef]
- Rehanek, M.; Karlin, D.G.; Bandte, M.; Al Kubrusli, R.; Nourinejhad Zarghani, S.; Candresse, T.; Büttner, C.; von Bargen, S. The Complex World of Emaraviruses—Challenges, Insights, and Prospects. Forests 2022, 13, 1868. [Google Scholar] [CrossRef]
- Domingo, E.; Escarmís, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuán, R. Extremely high mutation rate of a hammerhead viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef] [Green Version]
- Nourinejhad Zarghani, S.; Shams-Bakhsh, M.; Zand, N.; Sokhandan-Bashir, N.; Pazhouhandeh, M. Genetic analysis of Iranian population of Potato leafroll virus based on ORF0. Virus Genes. 2012, 45, 567–574. [Google Scholar] [CrossRef]
- Nourinejhad Zarghani, S.; Shams-Bakhsh, M.; Bashir, N.S.; Wetzel, T. Molecular Characterization of Whole Genomic RNA2 From Iranian Isolates of Grapevine Fanleaf Virus. J. Phytopathol. 2013, 161, 419–425. [Google Scholar] [CrossRef]
- Wang, Y.; Gaba, V.; Yang, J.; Palukaitis, P.; Gal-On, A. Characterization of Synergy Between Cucumber mosaic virus and Potyviruses in Cucurbit Hosts. Phytopathology 2002, 92, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Wintermantel, W.M. Co-infection of beet mosaic virus with beet yellowing viruses leads to increased symptom expression on sugar beet. Plant Dis. 2005, 89, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.F.; Bowen, K.L. Synergistic disease in pepper caused by the mixed infection of cucumber mosaic virus and Pepper mottle virus. Phytopathology 2006, 96, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Untiveros, M.; Fuentes, S.; Salazar, L.F. Synergistic interaction of sweet potato chlorotic stunt virus (Crinivirus) with carla-, cucumo-, ipomo-, and potyviruses infecting sweet potato. Plant Dis. 2007, 91, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Syller, J. Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol. Plant Pathol. 2012, 13, 204–216. [Google Scholar] [CrossRef]
- Moreno, A.B.; López-Moya, J.J. When viruses play team sports: Mixed infections in plants. Phytopathology 2020, 110, 29–48. [Google Scholar] [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Henderson, W.W.; Monroe, M.C.; St Jeor, S.C.; Thayer, W.P.; Rowe, J.E.; Peters, C.J.; Nichol, S.T. Naturally occurring Sin Nombre virus genetic reassortants. Virology 1995, 214, 602–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile, A.; Alonso-Prados, J.L.; Aranda, M.A.; Bernal, J.J.; Malpica, J.M.; Garcia-Arenal, F. Genetic exchange by recombination or reassortment is infrequent in natural populations of a tripartite RNA plant virus. J. Virol. 1997, 71, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Schnippenkoetter, W.; Martin, D.; Willment, J.; Rybicki, E. Forced recombination between distinct strains of maize streak virus. J. Gen. Virol. 2001, 82, 3081–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, D.N.; Martin, D.P.; Van der Walt, E.; Dent, K.; Varsani, A.; Rybicki, E.P. Maize streak virus: An old and complex ‘emerging’pathogen. Mol. Plant Pathol. 2010, 11, 1–12. [Google Scholar] [CrossRef]
- Butković, A.; González, R. A brief view of factors that affect plant virus evolution. Front. Virol. 2022, 2, 994057. [Google Scholar] [CrossRef]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Pagán, I. The diversity, evolution and epidemiology of plant viruses: A phylogenetic view. Infect. Genet. Evol. 2018, 65, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Mechanisms of plantvirus evolution. Annu. Rev. Phytopathol. 1997, 35, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.A.C.; Vazquez-Iglesias, I.; McGreig, S.; Fox, A.; Gibbs, A.J. Genomic High Plains wheat mosaic virus sequences from Australia: Their phylogenetics and evidence for emaravirus recombination and reassortment. Viruses 2023, 15, 401. [Google Scholar] [CrossRef]
- Danesh-Amuz, S.; Rakhshandehroo, F.; Rezaee, S. Prevalence and genetic diversity of fig mosaic virus isolates infecting fig tree in Iran. Acta Virol. 2014, 58, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Walia, J.J.; Willemsen, A.; Elci, E.; Caglayan, K.; Falk, B.W.; Rubio, L. Genetic variation and possible mechanisms driving the evolution of worldwide Fig mosaic virus isolates. Phytopathology 2014, 104, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.; Tzanetakis, I.E. Population structure, evolution and detection of blackberry leaf mottle-associated virus, an emerging emaravirus. Plant Pathol. 2019, 68, 775–782. [Google Scholar] [CrossRef]
- Rumbou, A.; Candresse, T.; von Bargen, S.; Buttner, C. Next-generation sequencing reveals a novel emaravirus in diseased maple trees from a german urban forest. Front. Microbiol. 2020, 11, 621179. [Google Scholar] [CrossRef]
- Dong, L.; Lemmetty, A.; Latvala, S.; Samuilova, O.; Valkonen, J.P.T. Occurrence and genetic diversity of raspberry leaf blotch virus (RLBV) infecting cultivated and wild Rubus species in Finland. Ann. Appl. Biol. 2016, 168, 122–132. [Google Scholar] [CrossRef]
- Jevremović, D.; Leposavić, A.; Paunović, S.A. Genetic diversity of raspberry leaf blotch emaravirus in red raspberries from Serbia. Span. J. Agric. Res. 2019, 17, e1004. [Google Scholar] [CrossRef]
- Verchot, J.; Herath, V.; Jordan, R.; Hammond, J. Genetic diversity among rose rosette virus isolates: A roadmap towards studies of gene function and pathogenicity. Pathogens 2023, 12, 707. [Google Scholar] [CrossRef]
- Guo, J.; Wang, Y.; Wang, G.; Hong, J.; Yang, Z.; Bai, J.; Hong, N. Molecular characteristics of jujube yellow mottle-associated virus infecting jujube (Ziziphus jujuba Mill.) Grown at Aksu in Xinjiang of China. Viruses 2020, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Usugi, T.; Tomitaka, Y.; Shimomoto, Y.; Takeuchi, S.; Kadono, F.; Yanagisawa, H.; Chiaki, Y.; Tsuda, S. Perilla mosaic virus is a highly divergent emaravirus transmitted by Shevtchenkella sp. (Acari: Eriophyidae). Phytopathology 2020, 110, 1352–1361. [Google Scholar] [CrossRef]
- Roßbach, J.; Dieckmann, H.L.; Büttner, T.; Mühlbach, H.-P.; von Bargen, S.; Büttner, C. Genetic variability and phylogeny of european mountain ash ringspot-associated virus RNA3 and RNA4. Forests 2015, 6, 4072–4087. [Google Scholar] [CrossRef] [Green Version]
- Tatineni, S.; Hein, G.L. High Plains wheat mosaic virus: An enigmatic disease of wheat and corn causing the High Plains disease. Mol. Plant Pathol. 2021, 22, 1167–1179. [Google Scholar] [CrossRef]
- Jacquot, M.; Wallace, M.A.; Streicker, D.G.; Biek, R. Geographic range overlap rather than phylogenetic distance explains rabies virus transmission among closely related bat species. Viruses 2022, 14, 2399. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Hughes, G.J.; Rupprecht, C.E. Phylogenetic relationships of seven previously unclassified viruses within the family Rhabdoviridae using partial nucleoprotein gene sequences. J. Gen. Virol. 2006, 87, 2323–2331. [Google Scholar] [CrossRef]
- Pappu, H.R.; du Toit, L.J.; Schwartz, H.F.; Mohan, S.K. Sequence diversity of the nucleoprotein gene of iris yellow spot virus (genus Tospovirus, family Bunyaviridae) isolates from the western region of the United States. Arch. Virol. 2006, 151, 1015–1025. [Google Scholar] [CrossRef]
- Herath, V.; Romay, G.; Urrutia, C.D.; Verchot, J. Family level phylogenies reveal relationships of plant viruses within the order Bunyavirales. Viruses 2020, 12, 1010. [Google Scholar] [CrossRef]
- Šantak, M.; Matić, Z. The role of nucleoprotein in immunity to human negative-stranded rna viruses—not just another brick in the viral nucleocapsid. Viruses 2022, 14, 521. [Google Scholar]
- Mielke-Ehret, N.; Muhlbach, H.P. Emaravirus: A novel genus of multipartite, negative strand RNA plant viruses. Viruses 2012, 4, 1515–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boom, R.; Sol, C.; Salimans, M.; Jansen, C.; Wertheim-van Dillen, P.; Van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Elbeaino, T.; Whitfield, A.; Sharma, M.; Digiaro, M. Emaravirus-specific degenerate PCR primers allowed the identification of partial RNA-dependent RNA polymerase sequences of Maize red stripe virus and Pigeonpea sterility mosaic virus. J. Virol. Methods 2013, 188, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Di Bello, P.L.; Ho, T.; Tzanetakis, I.E. The evolution of emaraviruses is becoming more complex: Seven segments identified in the causal agent of Rose rosette disease. Virus Res. 2015, 210, 241–244. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.6, 2004. Distributed by the author; Department of Genome Sciences, University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020, 7, veaa087. [Google Scholar] [CrossRef]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- French, N.; Yu, S.; Biggs, P.; Holland, B.; Fearnhead, P.; Binney, B.; Fox, A.; Grove-White, D.; Leigh, J.W.; Miller, W.; et al. Evolution of Campylobacter species in New Zealand. In Campylobacter Ecology and Evolution; Sheppard, S.K., Méric, G., Eds.; Caister Academic Press: Norfolk, England, 2014; pp. 221–240. [Google Scholar]
- Clement, M.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K. TCS: Estimating gene genealogies. Parallel Distrib. Process. Symp. Int. Proc. 2002, 2, 184. [Google Scholar]
- Hull, R. Matthews’ Plant Virology; Academic Press: San Diego, CA, USA, 2002; Volume 1056, p. 1001. [Google Scholar]
- Elbeaino, T.; Digiaro, M.; Uppala, M.; Sudini, H. Deep sequencing of dsRNAs recovered from mosaic-diseased pigeonpea reveals the presence of a novel emaravirus: Pigeonpea sterility mosaic virus 2. Arch. Virol. 2015, 160, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Elbeaino, T.; Digiaro, M.; Martelli, G.P. Complete nucleotide sequence of four RNA segments of fig mosaic virus. Arch. Virol. 2009, 154, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Karlin, D.G.; Lu, Y.; Wright, K.; Chen, J.; MacFarlane, S. Experimental and bioinformatic evidence that raspberry leaf blotch emaravirus P4 is a movement protein of the 30K superfamily. J. Gen. Virol. 2013, 94, 2117–2128. [Google Scholar] [CrossRef]
- McGavin, W.J.; Mitchell, C.; Cock, P.J.A.; Wright, K.M.; MacFarlane, S.A. Raspberry leaf blotch virus, a putative new member of the genus Emaravirus, encodes a novel genomic RNA. J. Gen. Virol. 2012, 93, 430–437. [Google Scholar] [CrossRef]
- Reguera, J.; Weber, F.; Cusack, S. Bunyaviridae RNA polymerases (L-Protein) have an n-terminal, influenza-like endonuclease domain, essential for viral Cap-dependent transcription. PLoS Path. 2010, 6, e1001101. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Poch, O.; Delarue, M.; Bishop, D.; Bouloy, M. Rift Valley fever virus L segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994, 75, 1345–1352. [Google Scholar] [CrossRef]
- Kumar, S.; Subbarao, B.; Hallan, V. Molecular characterization of emaraviruses associated with pigeonpea sterility mosaic disease. Sci. Rep. 2017, 7, 11831. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.T.; Ten Hagen, K.G. Mucin-type O-glycosylation during development. J. Biol. Chem. 2013, 288, 6921–6929. [Google Scholar] [CrossRef] [Green Version]
- Stewart, L.R. Sequence diversity of wheat mosaic virus isolates. Virus Res. 2016, 213, 299–303. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Plants, viruses and the environment: Ecology and mutualism. Virology 2015, 479-480, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, C.; Landgraf, M.; Fernandez Colino, H.L.; von Bargen, S.; Bandte, M. Chapter 3-Virus diseases of forest and urban trees. In Forest Microbiology; Asiegbu, F.O., Kovalchuk, A., Eds.; Academic Press: Cambridge, MA, USA, 2023; Volume 3, pp. 61–97. [Google Scholar]
- Savory, F.R.; Varma, V.; Ramakrishnan, U. Identifying geographic hot spots of reassortment in a multipartite plant virus. Evol. Appl. 2014, 7, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Liu, H.; Yin, Y.; Chen, T.-C.; Yeh, S.-D.; Zhang, Z.; Dong, J. Full-length M and L RNA sequences of tospovirus isolate 2009-GZT, which causes necrotic ringspot on tomato in China. Arch. Virol. 2016, 161, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Kallinen, A.; Lindberg, I.; Tugume, A.; Valkonen, J. Detection, distribution, and genetic variability of European mountain ash ringspot-associated virus. Phytopathology 2009, 99, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Thekke-Veetil, T.; Polashock, J.J.; Marn, M.V.; Plesko, I.M.; Schilder, A.C.; Keller, K.E.; Martin, R.R.; Tzanetakis, I.E. Population structure of blueberry mosaic associated virus: Evidence of reassortment in geographically distinct isolates. Virus Res. 2015, 201, 79–84. [Google Scholar] [CrossRef]
- Nourinejhad Zarghani, S.; Dupuis-Maguiraga, L.; Bassler, A.; Wetzel, T. Mapping of the exchangeable and dispensable domains of the RNA 2-encoded 2AHP protein of arabis mosaic nepovirus. Virology 2014, 458–459, 106–113. [Google Scholar] [CrossRef]
- Sokhandan Bashir, N.; Nourinejhad Zarghani, S.; Hejazi, M.S. Diversity of grapevine fanleaf virus isolates from Iran. Virus Res. 2007, 128, 144–148. [Google Scholar] [CrossRef]
- Novella, I.S.; Elena, S.F.; Moya, A.; Domingo, E.; Holland, J.J. Size of genetic bottlenecks leading to virus fitness loss is determined by mean initial population fitness. J. Virol. 1995, 69, 2869–2872. [Google Scholar] [CrossRef]
- Bergstrom, C.T.; McElhany, P.; Real, L.A. Transmission bottlenecks as determinants of virulence in rapidly evolving pathogens. Proc. Natl. Acad. Sci. USA 1999, 96, 5095–5100. [Google Scholar] [CrossRef]
- Tsompana, M.; Abad, J.; Purugganan, M.; Moyer, J.W. The molecular population genetics of the tomato spotted wilt virus (TSWV) genome. Mol. Ecol. 2005, 14, 53–66. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Restrictotype | Position of AanI Site on cRNA3 | No. Cut 1 | No. Fr. 2 | Size of Fragments (bp) | Frequency of Restrictotypes (%) 3 | ||
|---|---|---|---|---|---|---|---|
| S | F | T | |||||
| Type I |  | 0 | 1 | 1587 to 1589 | 1.2 | 6.1 | 7.3 |
| Type II |  | 1 | 2 | 1474, and 115 | 0.0 | 1.2 | 1.2 |
| Type III |  | 1 | 2 | 962, and 627 | 2.4 | 3.7 | 6.1 |
| Type IV |  | 2 | 3 | 627, 521, and 442 | 0.0 | 4.9 | 4.9 |
| Type V |  | 3 | 4 | 627, 515, 266, and 182 | 1.2 | 2.4 | 3.7 |
| Type VI |  | 3 | 4 | 627, 442, 338, and 182 | 3.7 | 1.2 | 4.9 |
| Type VII |  | 4 | 5 | 627, 442, 264, 182, and 73 | 32.9 | 39.0 | 72.0 |
| Origin of Isolates | Genomic Region | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| RNA3 | RNA4 | Partial RNA1 | |||||||
| 5′ UTR | ORF3 | 3′ UTR | 5′ UTR | ORF4 | 3′ UTR | 5′ UTR | ORF1 | ||
| Finland | nt | 95–100 | 88.9–100 | 80.8–98.8 | 97.6–100 | 95.9–99.9 | 82.4–100 | 96.5–100 | 85.2–99.7 |
| aa | 88.4–100 | 96.9–100 | 96.6–100 | ||||||
| Sweden | nt | 94–100 | 89.4–99.6 | 80–98.8 | 100 | 95.9–99.9 | 80–98.8 | 97.6–100 | 87.4–98.7 |
| aa | 91.8–100 | 97.5–100 | 97.1–99.7 | ||||||
| Genomic Regions | Geographical Group | m | S | Tajima’s D | Fu and Li’s D | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RNA3 | ||||||||||
| Sweden | 9 | 275 | 0.06347 ± (0.02651) | 0.04735 ± (0.01764) | 0.11984 | 0.02115 | 0.176 | −1.322703 | −1.56810 | |
| Finland | 18 | 298 | 0.05435 ± (0.01881) | 0.04207 ± (0.01138) | 0.11180 | 0.02432 | 0.217 | −0.961799 | −0.47563 | |
| Total | 27 | 361 | 0.05876 ± (0.01844) | 0.04994 ± (0.00834) | 0.12843 | 0.02695 | 0.210 | −0.579852 | −0.60644 | |
| RNA4 | ||||||||||
| Sweden | 8 | 119 | 0.02948 ± (0.01286) | 0.03048 ± (0.00391) | 0.09485 | 0.00542 | 0.057 | 0.186391 | −0.14740 | |
| Finland | 18 | 141 | 0.02633 ± (0.00929) | 0.02479 ± (0.00237) | 0.08977 | 0.00418 | 0.047 | −0.246562 | −0.29650 | |
| Total | 26 | 190 | 0.03198 ± (0.01032) | 0.02738 ± (0.00187) | 0.09406 | 0.00460 | 0.049 | −0.568813 | −0.66359 | |
| Partial RNA1 | ||||||||||
| Sweden | 8 | 242 | 0.05930 ± (0.02564) | 0.04447 ± (0.01850) | 0.19665 | 0.00934 | 0.048 | −1.362813 | −1.50039 | |
| Finland | 18 | 417 | 0.07702 ± (0.02673) | 0.08018 ± (0.00753) | 0.40730 | 0.01603 | 0.039 | 0.174721 | 0.10686 | |
| Total | 26 | 451 | 0.07509 ± (0.02390) | 0.07777 ± (0.00663) | 0.39134 | 0.01554 | 0.040 | 0.153180 | −0.04648 | |
| Genomic Region | Populations | KST* | p-Value | Z* | p-Value | Snn | p-Value | Hs | p-Value | FST |
|---|---|---|---|---|---|---|---|---|---|---|
| RNA3 | S. vs. F | 0.07092 | 0.0000 *** | 4.51006 | 0.0000 *** | 0.85185 | 0.0010 ** | 0.99545 | 0.5330 ns | 0.22767 |
| RNA4 | S. vs. F | 0.02288 | 0.0080 ** | 4.63422 | 0.0050 ** | 0.88462 | 0.0030 ** | 1.00000 | 1.0000 ns | 0.06408 |
| RNA1 | S. vs. F | 0.03890 | 0.0000 *** | 4.44027 | 0.0000 *** | 0.92308 | 0.0010 * | 1.00000 | 1.0000 ns | 0.23698 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nourinejhad Zarghani, S.; Al Kubrusli, R.; Iancev, S.; Jalkanen, R.; Büttner, C.; von Bargen, S. Molecular Population Genetics of Aspen Mosaic-Associated Virus in Finland and Sweden. Viruses 2023, 15, 1678. https://doi.org/10.3390/v15081678
Nourinejhad Zarghani S, Al Kubrusli R, Iancev S, Jalkanen R, Büttner C, von Bargen S. Molecular Population Genetics of Aspen Mosaic-Associated Virus in Finland and Sweden. Viruses. 2023; 15(8):1678. https://doi.org/10.3390/v15081678
Chicago/Turabian StyleNourinejhad Zarghani, Shaheen, Rim Al Kubrusli, Serghei Iancev, Risto Jalkanen, Carmen Büttner, and Susanne von Bargen. 2023. "Molecular Population Genetics of Aspen Mosaic-Associated Virus in Finland and Sweden" Viruses 15, no. 8: 1678. https://doi.org/10.3390/v15081678
APA StyleNourinejhad Zarghani, S., Al Kubrusli, R., Iancev, S., Jalkanen, R., Büttner, C., & von Bargen, S. (2023). Molecular Population Genetics of Aspen Mosaic-Associated Virus in Finland and Sweden. Viruses, 15(8), 1678. https://doi.org/10.3390/v15081678