
Update on the Phylodynamic and Genetic Variability of Marburg Virus

 ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
Phylodynamics
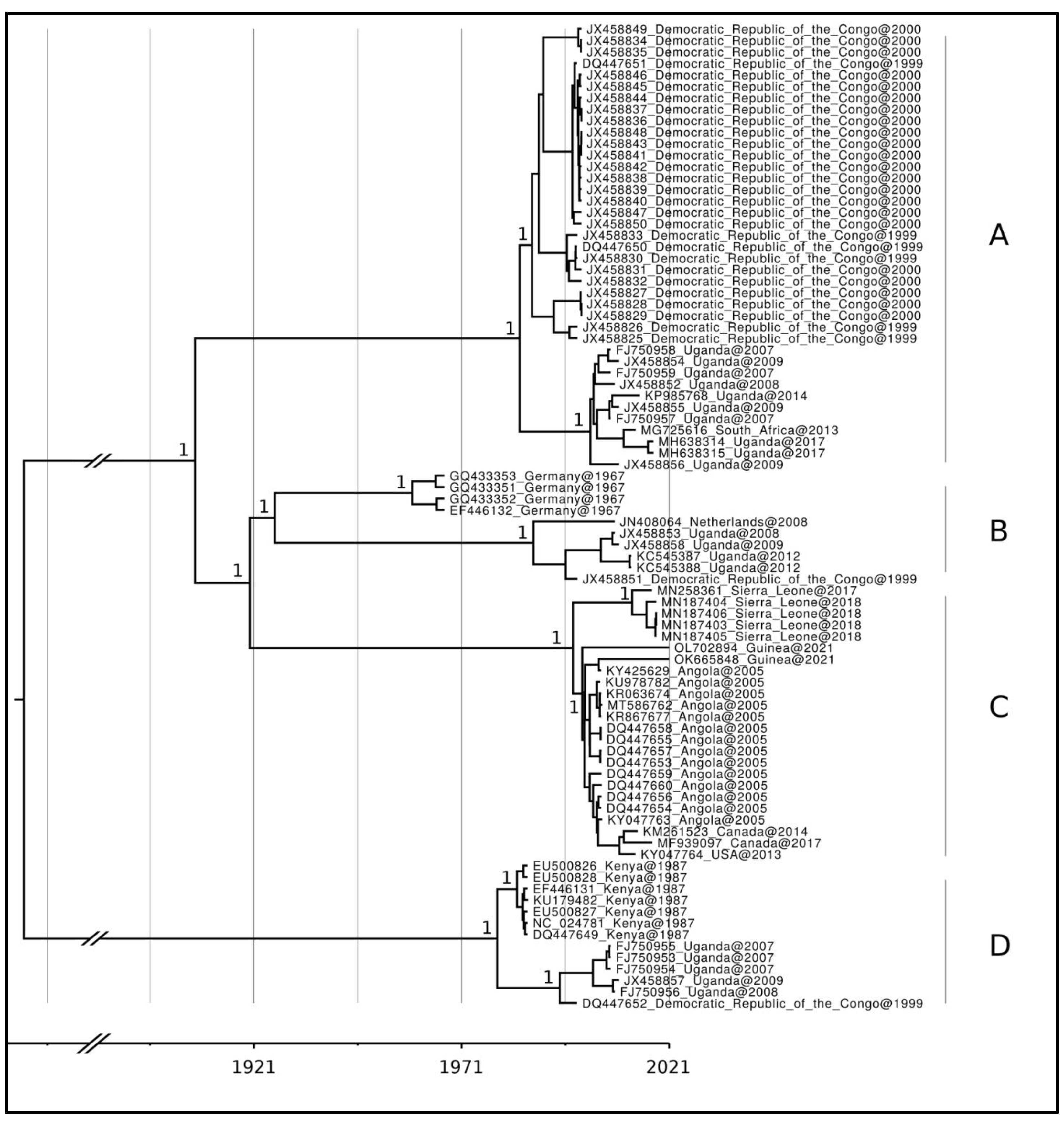
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tegally, H.; San, J.E.; Cotten, M.; Moir, M.; Tegomoh, B.; Mboowa, G.; Martin, D.P.; Baxter, C.; Lambisia, A.W.; Diallo, A.; et al. The evolving SARS-CoV-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. Science 2022, 378, eabq5358. [Google Scholar] [CrossRef]
- Eneh, S.C.; Okonji, O.C.; Chiburoma, A.G.; Francisca Ogochukwu, O.; Tuwleh, L.; Gideon, I.; Okonji, E.F.; Bushabu, F.N.; Mgbere, O. Marburg virus disease amid COVID-19 in West Africa: An emerging and re-emerging zoonotic epidemic threat, future implications and way forward. Ther. Adv. Infect. Dis. 2023, 10, 20499361231168520. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T.; et al. Proposal for a revised taxonomy of the family Filoviridae: Classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asad, A.; Aamir, A.; Qureshi, N.E.; Bhimani, S.; Jatoi, N.N.; Batra, S.; Ochani, R.K.; Abbasi, M.K.; Tariq, M.A.; Diwan, M.N. Past and current advances in Marburg virus disease: A review. Infez. Med. 2020, 28, 332–345. [Google Scholar] [PubMed]
- Rudge, T.L., Jr.; Machesky, N.J.; Sankovich, K.A.; Lemmon, E.E.; Badorrek, C.S.; Overman, R.; Niemuth, N.A.; Anderson, M.S. Assays for the Evaluation of the Immune Response to Marburg and Ebola Sudan Vaccination-Filovirus Animal Nonclinical Group Anti-Marburg Virus Glycoprotein Immunoglobulin G Enzyme-Linked Immunosorbent Assay and a Pseudovirion Neutralization Assay. Vaccines 2022, 10, 1211. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Disease Outbreak News—Marburg Virus Disease—Equatorial Guinea. 2023. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON459#:~:text=On%2013%20February%202023%2C%20the,virus%20by%20real%2Dtime%20polymerase (accessed on 1 May 2023).
- República de Guinea Ecuatorial; Ministerio de Sanidad y Bienestar Social. Actualización de Datos Epidemiológicos. Available online: https://guineasalud.org/ (accessed on 1 May 2023).
- World Health Organization. Disease Outbreak News—Marburg Virus Disease—Equatorial Guinea and the United Republic of Tanzania. 2023. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON467 (accessed on 1 July 2023).
- Zhao, F.; He, Y.; Lu, H. Marburg virus disease: A deadly rare virus is coming. Biosci. Trends 2022, 16, 312–316. [Google Scholar] [CrossRef]
- National Library of Medicine—National Center for Biotechnology Information. NCBI Virus. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ (accessed on 15 June 2023).
- Scarpa, F.; Sanna, D.; Benvenuto, D.; Borsetti, A.; Azzena, I.; Casu, M.; Fiori, P.L.; Giovanetti, M.; Maruotti, A.; Ceccarelli, G.; et al. Genetic and Structural Data on the SARS-CoV-2 Omicron BQ.1 Variant Reveal Its Low Potential for Epidemiological Expansion. Int. J. Mol. Sci. 2022, 23, 15264. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- FigTree. Software for Molecular Evolution, Phylogeny and Epidemiology. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 15 July 2023).
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, e214. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 645 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugosa, B.; Vujosevic, D.; Ciccozzi, M.; Valli, M.B.; Capobianchi, M.R.; Presti, A.L.; Cella, E.; Giovanetti, M.; Lai, A.; Angeletti, S.; et al. Genetic diversity of the haemagglutinin (HA) of human influenza A (H1N1) virus in Montenegro: Focus on its origin and evolution. J. Med. Virol. 2016, 88, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, J.H. Ebolavirus and marburgvirus infections. In Harrison’s Principles of Internal Medicine, 20th ed.; Jameson, J.L., Fauci, A.S., Kasper, D.L., Hauser, S.L., Longo, D.L., Loscalzo, J., Eds.; McGraw—Hill Education: Columbus, OH, USA, 2018; Volume 2, pp. 1509–1515. [Google Scholar]
- Mire, C.E.; Geisbert, J.B.; Borisevich, V.; Fenton, K.A.; Agans, K.N.; Flyak, A.I.; Deer, D.J.; Steinkellner, H.; Bohorov, O.; Bohorova, N.; et al. Therapeutic treatment of Marburg and Ravn virus infection in nonhuman primates with a human monoclonal antibody. Sci. Transl. Med. 2017, 9, eaai8711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espy, N.; Nagle, E.; Pfeffer, B.; Garcia, K.; Chitty, A.J.; Wiley, M.; Sanchez-Lockhart, M.; Bavari, S.; Warren, T.; Palacios, G. T-705 induces lethal mutagenesis in Ebola and Marburg populations in macaques. Antivir. Res. 2019, 170, 104529. [Google Scholar] [CrossRef]
- Warren, T.K.; Whitehouse, C.A.; Wells, J.; Welch, L.; Charleston, J.S.; Heald, A.; Nichols, D.K.; Mattix, M.E.; Palacios, G.; Kugleman, J.R.; et al. Delayed time-to-treatment of an antisense morpholino oligomer is effective against lethal Marburg virus infection in cynomolgus macaques. PLoS Negl. Trop. Dis. 2016, 10, e0004456. [Google Scholar] [CrossRef] [Green Version]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Ströher, U.; et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [Green Version]
- Knust, B.; Schafer, I.J.; Wamala, J.; Nyakarahuka, L.; Okot, C.; Shoemaker, T.; Dodd, K.; Gibbons, A.; Balinandi, S.; Tumusiime, A.; et al. Multidistrict outbreak of Marburg virus disease—Uganda, 2012. J. Infect. Dis. 2015, 212, S119–S128. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Locci, C.; Angeletti, S.; Maruotti, A.; Ceccarelli, G.; Casu, M.; Fiori, P.L.; et al. Genetic Variability of the Monkeypox Virus Clade IIb B.1. J. Clin. Med. 2022, 11, 6388. [Google Scholar] [CrossRef]
- Carroll, S.A.; Towner, J.S.; Sealy, T.K.; McMullan, L.K.; Khristova, M.L.; Burt, F.J.; Swanepoel, R.; Rollin, P.E.; Nichol, S.T. Molecular evolution of viruses of the family Filoviridae based on 97 whole-genome sequences. J. Virol. 2013, 87, 2608–2616. [Google Scholar] [CrossRef] [Green Version]
- Zehender, G.; Sorrentino, C.; Veo, C.; Fiaschi, L.; Gioffrè, S.; Ebranati, E.; Tanzi, E.; Ciccozzi, M.; Lai, A.; Galli, M. Distribution of Marburg virus in Africa: An evolutionary approach. Infect. Genet. Evol. 2016, 44, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Bausch, D.G.; Nichol, S.T.; Muyembe-Tamfum, J.J.; Borchert, M.; Rollin, P.E.; Sleurs, H.; Campbell, P.; Tshioko, F.K.; Roth, C.; Colebunders, R.; et al. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N. Engl. J. Med. 2006, 355, 909–919. [Google Scholar] [CrossRef]
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Taylor, J.; George, K.S.; Griesemer, S.B.; Ghedin, E.; Sengamalay, N.A.; Spiro, D.J.; et al. Correction: Stochastic Processes Are Key Determinants of Short-Term Evolution in Influenza a Virus. PLoS Pathog. 2006, 2, e138. [Google Scholar] [CrossRef]
- Armstrong, G.L.; MacCannell, D.R.; Taylor, J.; Carleton, H.A.; Neuhaus, E.B.; Bradbury, R.S.; Posey, J.E.; Gwinn, M. Pathogen Genomics in Public Health. N. Engl. J. Med. 2019, 381, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Giovanetti, M.; Benvenuto, D.; Coradduzza, E.; Alexiev, I.; Casu, M.; Fiori, P.L.; et al. Update on the Phylodynamics of SADS-CoV. Life 2021, 11, 820. [Google Scholar] [CrossRef]
- Duault, H.; Durand, B.; Canini, L. Methods Combining Genomic and Epidemiological Data in the Reconstruction of Transmission Trees: A Systematic Review. Pathogens 2022, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Jeggo, M. The One Health Approach—Why Is It So Important? Trop. Med. Infect. Dis. 2019, 4, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling-Hu, T.; Rios-Guzman, E.; Lorenzo-Redondo, R.; Ozer, E.A.; Hultquist, J.F. Challenges and Opportunities for Global Genomic Surveillance Strategies in the COVID-19 Era. Viruses 2022, 14, 2532. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpa, F.; Bazzani, L.; Giovanetti, M.; Ciccozzi, A.; Benedetti, F.; Zella, D.; Sanna, D.; Casu, M.; Borsetti, A.; Cella, E.; et al. Update on the Phylodynamic and Genetic Variability of Marburg Virus. Viruses 2023, 15, 1721. https://doi.org/10.3390/v15081721
Scarpa F, Bazzani L, Giovanetti M, Ciccozzi A, Benedetti F, Zella D, Sanna D, Casu M, Borsetti A, Cella E, et al. Update on the Phylodynamic and Genetic Variability of Marburg Virus. Viruses. 2023; 15(8):1721. https://doi.org/10.3390/v15081721
Chicago/Turabian StyleScarpa, Fabio, Liliana Bazzani, Marta Giovanetti, Alessandra Ciccozzi, Francesca Benedetti, Davide Zella, Daria Sanna, Marco Casu, Alessandra Borsetti, Eleonora Cella, and et al. 2023. "Update on the Phylodynamic and Genetic Variability of Marburg Virus" Viruses 15, no. 8: 1721. https://doi.org/10.3390/v15081721
APA StyleScarpa, F., Bazzani, L., Giovanetti, M., Ciccozzi, A., Benedetti, F., Zella, D., Sanna, D., Casu, M., Borsetti, A., Cella, E., Pascarella, S., Maruotti, A., & Ciccozzi, M. (2023). Update on the Phylodynamic and Genetic Variability of Marburg Virus. Viruses, 15(8), 1721. https://doi.org/10.3390/v15081721