
Vaccines’ New Era-RNA Vaccine

 , , , , , and
, , , , , and
Abstract
:1. Introduction
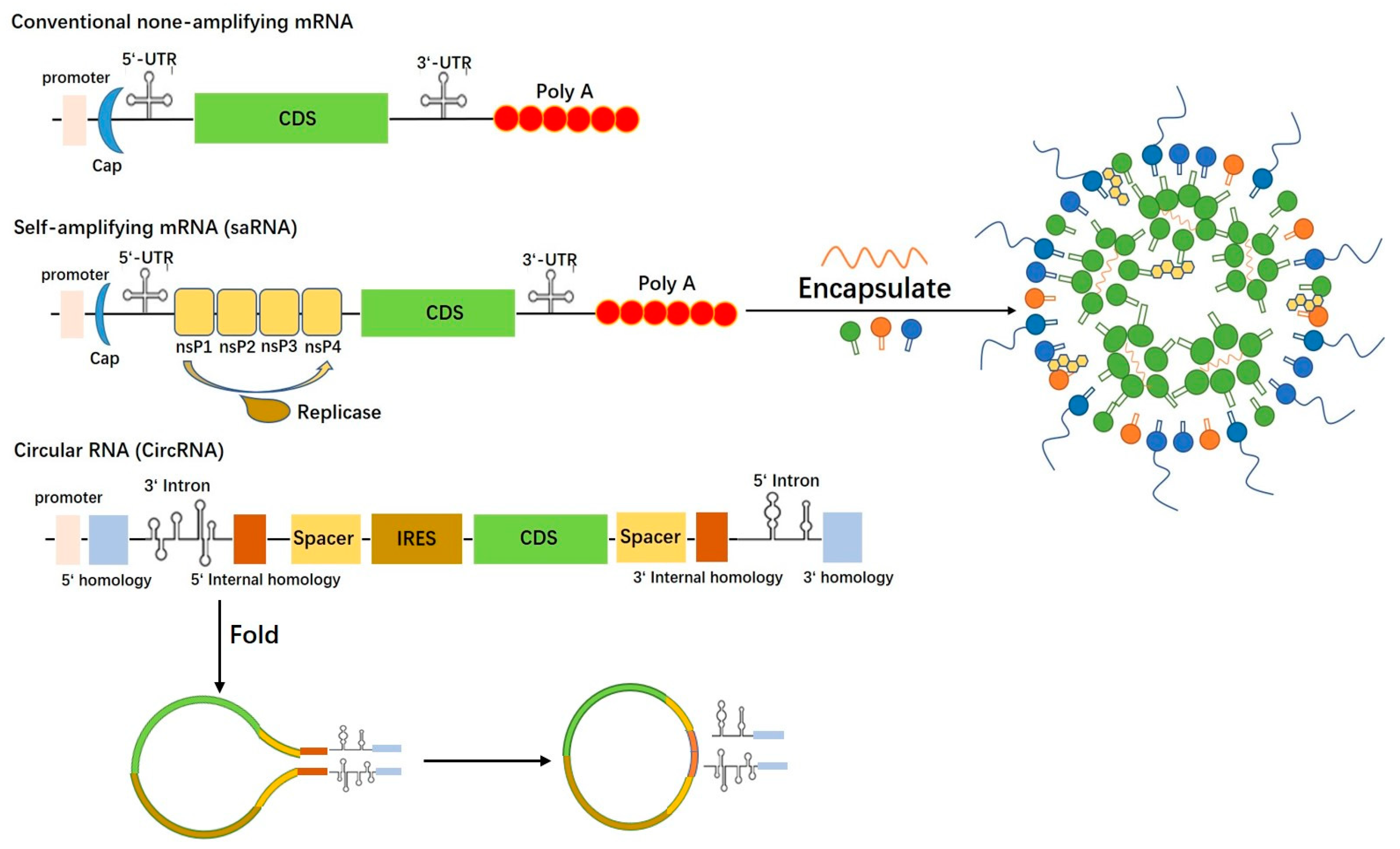
1.1. Conventional mRNA Vaccine
1.2. CircRNA Vaccine
1.3. saRNA Vaccine
2. RNA Vaccines for Infectious Diseases
2.1. Coronavirus
2.2. Flavivirus
2.3. Influenza Virus
2.4. Respiratory Syncytial Virus (RSV)
3. RNA Vaccine Delivery System
3.1. Lipid Nanoparticles (LNP)
3.2. Extracellular Vesicles (EVs)
3.3. Other Delivery Systems (Protamine and Hydrogel)
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dolgin, E. The Tangled History of MRNA Vaccines. Nature 2021, 597, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A. BNT162b Vaccines Protect Rhesus Macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.-P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Peña, A.T.; Korzun, J. A Next-Generation Cleaved, Soluble HIV-1 Env Trimer, BG505 SOSIP. 664 Gp140, Expresses Multiple Epitopes for Broadly Neutralizing but Not Non-Neutralizing Antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Aoshi, T.; Ozasa, K.; Kusakabe, T.; Momota, M.; Haseda, Y.; Kobari, S.; Kuroda, E.; Kobiyama, K.; Coban, C. RNA Is an Adjuvanticity Mediator for the Lipid-Based Mucosal Adjuvant, Endocine. Sci. Rep. 2016, 6, 29165. [Google Scholar] [CrossRef]
- Swaminathan, G.; Thoryk, E.A.; Cox, K.S.; Smith, J.S.; Wolf, J.J.; Gindy, M.E.; Casimiro, D.R.; Bett, A.J. A Tetravalent Sub-unit Dengue Vaccine Formulated with Ionizable Cationic Lipid Nanoparticle induces Significant Immune Responses in Rodents and Non-Human Primates. Sci. Rep. 2016, 6, 34215. [Google Scholar] [CrossRef]
- Alameh, M.G.; Tombácz, I.; Bettini, E.; Lederer, K.; Sittplangkoon, C.; Wilmore, J.R.; Gaudette, B.T.; Soliman, O.Y.; Pine, M.; Hicks, P.; et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 2021, 54, 2877–2892.e7. [Google Scholar] [CrossRef]
- Matsumoto, M.; Seya, T. TLR3: Interferon induction by double-stranded RNA including poly(I:C). Adv. Drug Deliv. Rev. 2008, 60, 805–812. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Li, C.; Lee, A.; Grigoryan, L.; Arunachalam, P.S.; Scott, M.K.D.; Trisal, M.; Wimmers, F.; Sanyal, M.; Weidenbacher, P.A.; Feng, Y.; et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat. Immunol. 2022, 23, 543–555. [Google Scholar] [CrossRef]
- Esparza, J.; Lederman, S.; Nitsche, A.; Damaso, C.R. Early Smallpox Vaccine Manufacturing in the United States: Introduction of the “Animal Vaccine” in 1870, Establishment of “Vaccine Farms”, and the Beginnings of the Vaccine Industry. Vaccine 2020, 38, 4773–4779. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; et al. Vaccine Breakthrough Infections with SARS-CoV-2 Variants. N. Engl. J. Med. 2021, 384, 2212–2218. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Xu, Q. Current Developments and Challenges of MRNA Vaccines. Annu. Rev. Biomed. Eng. 2022, 24, 85–109. [Google Scholar] [CrossRef]
- Pang, Y.; Mao, C.; Liu, S. Encoding Activities of Non-Coding RNAs. Theranostics 2018, 8, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, M.; Zhang, L.; Huang, S.; Xiao, F.; Zou, L. Circular RNAs: Biomarkers of Cancer. Cancer Innov. 2022, 1, 197–206. [Google Scholar] [CrossRef]
- Zhao, W.; Dong, M.; Pan, J.; Wang, Y.; Zhou, J.; Ma, J.; Liu, S. Circular RNAs: A novel target among non–coding RNAs with potential roles in malignant tumors. Mol. Med. Rep. 2019, 20, 3463–3474. [Google Scholar] [CrossRef]
- Macejak, D.G.; Sarnow, P. Internal Initiation of Translation Mediated by the 5′ Leader of a Cellular MRNA. Nature 1991, 353, 90–94. [Google Scholar] [CrossRef]
- Chen, C.; Sarnow, P. Initiation of Protein Synthesis by the Eukaryotic Translational Apparatus on Circular RNAs. Science 1995, 268, 415–417. [Google Scholar] [CrossRef]
- AbouHaidar, M.G.; Venkataraman, S.; Golshani, A.; Liu, B.; Ahmad, T. Novel Coding, Translation, and Gene Expression of a Replicating Covalently Closed Circular RNA of 220 Nt. Proc. Natl. Acad. Sci. USA 2014, 111, 14542–14547. [Google Scholar] [CrossRef]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M. Circ-ZNF609 Is a Circular RNA That Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37.e9. [Google Scholar] [CrossRef]
- Xia, X.; Li, X.; Li, F.; Wu, X.; Zhang, M.; Zhou, H.; Huang, N.; Yang, X.; Xiao, F.; Liu, D. A Novel Tumor Suppressor Protein Encoded by Circular AKT3 RNA Inhibits Glioblastoma Tumorigenicity by Competing with Active Phosphoinositide-Dependent Kinase-1. Mol. Cancer 2019, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.-C.; Wong, C.-W.; Liang, P.-P.; Shi, M.; Cao, Y.; Rao, S.-T.; Tsui, S.K.-W.; Waye, M.M.-Y.; Zhang, Q.; Fu, W.-M. Translation of the Circular RNA Circβ-Catenin Promotes Liver Cancer Cell Growth through Activation of the Wnt Pathway. Genome Biol. 2019, 20, 84. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, X.; Zhang, M.; Yan, S.; Sun, C.; Xiao, F.; Huang, N.; Yang, X.; Zhao, K.; Zhou, H. Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. JNCI J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H. A Novel Protein Encoded by the Circular Form of the SHPRH Gene Suppresses Glioma Tumorigenesis. Oncogene 2018, 37, 1805–1814. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, K.; Xu, X.; Yang, Y.; Yan, S.; Wei, P.; Liu, H.; Xu, J.; Xiao, F.; Zhou, H. A Peptide Encoded by Circular Form of LINC-PINT Suppresses Oncogenic Transcriptional Elongation in Glioblastoma. Nat. Commun. 2018, 9, 4475. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR M6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.-L.; Wang, Y. Extensive Translation of Circular RNAs Driven by N6-Methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef]
- Fan, X.; Yang, Y.; Chen, C.; Wang, Z. Pervasive Translation of Circular RNAs Driven by Short IRES-like Elements. Nat. Commun. 2022, 13, 3751. [Google Scholar] [CrossRef]
- Obi, P.; Chen, Y.G. The Design and Synthesis of Circular RNAs. Methods 2021, 196, 85–103. [Google Scholar] [CrossRef]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide Synthesis Using T7 RNA Polymerase and Synthetic DNA Templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef]
- Gholamalipour, Y.; Karunanayake Mudiyanselage, A.; Martin, C.T. 3′ End Additions by T7 RNA Polymerase Are RNA Self-Templated, Distributive and Diverse in Character—RNA-Seq Analyses. Nucleic Acids Res. 2018, 46, 9253–9263. [Google Scholar] [CrossRef]
- Lohman, G.J.S.; Tabor, S.; Nichols, N.M. Current Protocols in Molecular Biology—DNA ligases. Curr. Protoc. Mol. Biol. 2011, 3.14.1–3.14.7. [Google Scholar] [CrossRef]
- Moore, M.J.; Query, C.C. Joining of RNAs by splinted ligation. Methods Enzymol. 2000, 317, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbeck, O.C.; Gumport, R.I. T4 RNA ligase. Enzyme 1982, 15, 31–58. [Google Scholar]
- Petkovic, S.; Müller, S. RNA circularization strategies in vivo and in vitro. Nucleic Acids Res. 2015, 43, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Yi, Z.; Shen, Y.; Lin, L.; Chen, F.; Xu, Y.; Wu, Z.; Tang, H.; Zhang, X.; Tian, F.; et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022, 185, 1728–1744.e16. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Kodama, A.; Abe, H. Circular RNAs, methods and protocols—Preparation of circRNA in vitro. Methods Mol. Biol. 2018, 1724, 181–192. [Google Scholar]
- Puttaraju, M.; Been, M. Group I Permuted Intron-Exon (PIE) Sequences Self-Splice to Produce Circular Exons. Nucleic Acids Res. 1992, 20, 5357–5364. [Google Scholar] [CrossRef]
- Ford, E.; Ares Jr, M. Synthesis of Circular RNA in Bacteria and Yeast Using RNA Cyclase Ribozymes Derived from a Group I Intron of Phage T4. Proc. Natl. Acad. Sci. USA 1994, 91, 3117–3121. [Google Scholar] [CrossRef] [PubMed]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering Circular RNA for Potent and Stable Translation in Eukaryotic Cells. Nat. Commun. 2018, 9, 2629. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, S.; Hakim-Zargar, M.; Carlson, D.; Jarrell, K. Use of an Engineered Ribozyme to Produce a Circular Human Exon. Nucleic Acids Res. 1997, 25, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wei, H.; Zhang, K.; Li, Z.; Wei, T.; Tang, C.; Yang, Y.; Wang, Z. A Flexible, Efficient, and Scalable Platform to Produce Circular RNAs as New Therapeutics. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ballesteros-Briones, M.C.; Silva-Pilipich, N.; Herrador-Cañete, G.; Vanrell, L.; Smerdou, C. A New Generation of Vaccines Based on Alphavirus Self-Amplifying RNA. Curr. Opin. Virol. 2020, 44, 145–153. [Google Scholar] [CrossRef]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to MRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef]
- De Alwis, R.; Gan, E.S.; Chen, S.; Leong, Y.S.; Tan, H.C.; Zhang, S.L.; Yau, C.; Low, J.G.; Kalimuddin, S.; Matsuda, D. A Single Dose of Self-Transcribing and Replicating RNA-Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity in Mice. Mol. Ther. 2021, 29, 1970–1983. [Google Scholar] [CrossRef]
- Ljungberg, K.; Liljeström, P. Self-Replicating Alphavirus RNA Vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef]
- Aldon, Y.; McKay, P.F.; Herrero, J.M.; Vogel, A.B.; Lévai, R.; Maisonnasse, P.; Dereuddre-Bosquet, N.; Haas, H.; Fábián, K.; Le Grand, R. Immunogenicity of Stabilized HIV-1 Env Trimers Delivered by Self-Amplifying MRNA. Mol. Ther. Nucleic Acids 2021, 25, 483–493. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö. A Trans-Amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef]
- Pollock, K.M.; Cheeseman, H.M.; Szubert, A.J.; Libri, V.; Boffito, M.; Owen, D.; Bern, H.; O’Hara, J.; McFarlane, L.R.; Lemm, N.-M. Safety and Immunogenicity of a Self-Amplifying RNA Vaccine against COVID-19: COVAC1, a Phase I, Dose-Ranging Trial. EClinicalMedicine 2022, 44, 101262. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Crosby, E.J.; Force, J.; Osada, T.; Hobeika, A.C.; Hartman, Z.C.; Berglund, P.; Smith, J.; Lyerly, H.K. Clinical Trials of Self-Replicating RNA-Based Cancer Vaccines. Cancer Gene Ther. 2023, 30, 803–811. [Google Scholar] [CrossRef]
- Available online: https://covid19.who.int/ (accessed on 22 June 2023).
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Qamar, S.; Tekin, A.; Singh, R.; Kashyap, R. Vaccine Development Throughout History. Cureus 2021, 13, e16635. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 MRNA Vaccine Design Enabled by Prototype Pathogen Preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E.; Frenck, R.W.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Canetti, M.; Barda, N.; Gilboa, M.; Indenbaum, V.; Mandelboim, M.; Gonen, T.; Asraf, K.; Weiss-Ottolenghi, Y.; Amit, S.; Doolman, R.; et al. Immunogenicity and Efficacy of Fourth BNT162b2 and MRNA1273 COVID-19 Vaccine Doses; Three Months Follow-Up. Nat. Commun. 2022, 13, 7711. [Google Scholar] [CrossRef] [PubMed]
- Accorsi, E.K.; Britton, A.; Fleming-Dutra, K.E.; Smith, Z.R.; Shang, N.; Derado, G.; Miller, J.; Schrag, S.J.; Verani, J.R. Association between 3 Doses of MRNA COVID-19 Vaccine and Symptomatic Infection Caused by the SARS-CoV-2 Omicron and Delta Variants. JAMA 2022, 327, 639–651. [Google Scholar] [CrossRef]
- Muik, A.; Lui, B.G.; Wallisch, A.-K.; Bacher, M.; Mühl, J.; Reinholz, J.; Ozhelvaci, O.; Beckmann, N.; Güimil Garcia, R.d.C.; Poran, A.; et al. Neutralization of SARS-CoV-2 Omicron by BNT162b2 MRNA Vaccine–Elicited Human Sera. Science 2022, 375, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, Y.; Liu, X.; Hou, F.; Cai, R.; Yu, Z.; Liu, F.; Yang, G.; Ding, J.; Xu, J.; et al. Heterologous Boost with MRNA Vaccines against SARS-CoV-2 Delta/Omicron Variants Following an Inactivated Whole-Virus Vaccine. Antiviral Res. 2023, 212, 105556. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/vaccines-blood-biologics/qa-spikevax-covid-19-vaccine-mrna (accessed on 24 June 2023).
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-Amplifying RNA SARS-CoV-2 Lipid Nanoparticle Vaccine Candidate Induces High Neutralizing Antibody Titers in Mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef]
- Szubert, A.J.; Pollock, K.M.; Cheeseman, H.M.; Alagaratnam, J.; Bern, H.; Bird, O.; Boffito, M.; Byrne, R.; Cole, T.; Cosgrove, C.A.; et al. COVAC1 Phase 2a Expanded Safety and Immunogenicity Study of a Self-Amplifying RNA Vaccine against SARS-CoV-2. eClinicalMedicine 2023, 56, 101823. [Google Scholar] [CrossRef]
- Knipe, D.; Howley, P.; Griffin, D.; Lamb, R.; Martin, M.; Roizman, B.; Straus, S. Fields Virology, Volumes 1 and 2; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1. [Google Scholar]
- Collins, M.H.; Metz, S.W. Progress and Works in Progress: Update on Flavivirus Vaccine Development. Clin. Ther. 2017, 39, 1519–1536. [Google Scholar] [CrossRef]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue Virus Pathogenesis: An Integrated View. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, L.; Vapalahti, O. Tick-Borne Encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Tuells, J.; Henao-Martínez, A.F.; Franco-Paredes, C. Yellow Fever: A Perennial Threat. Arch. Med. Res. 2022, 53, 649–657. [Google Scholar] [CrossRef]
- Essink, B.; Chu, L.; Seger, W.; Barranco, E.; Le Cam, N.; Bennett, H.; Faughnan, V.; Pajon, R.; Paila, Y.D.; Bollman, B. The Safety and Immunogenicity of Two Zika Virus MRNA Vaccine Candidates in Healthy Flavivirus Baseline Seropositive and Seronegative Adults: The Results of Two Randomised, Placebo-Controlled, Dose-Ranging, Phase 1 Clinical Trials. Lancet Infect. Dis. 2023, 23, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Bollman, B.; Nunna, N.; Bahl, K.; Hsiao, C.J.; Bennett, H.; Butler, S.; Foreman, B.; Burgomaster, K.E.; Aleshnick, M.; Kong, W.-P. An Optimized Messenger RNA Vaccine Candidate Protects Non-Human Primates from Zika Virus Infection. Npj Vaccines 2023, 8, 58. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O. The Global Distribution and Burden of Dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Roth, C.; Cantaert, T.; Colas, C.; Prot, M.; Casadémont, I.; Levillayer, L.; Thalmensi, J.; Langlade-Demoyen, P.; Gerke, C.; Bahl, K. A Modified MRNA Vaccine Targeting Immunodominant NS Epitopes Protects against Dengue Virus Infection in HLA Class I Transgenic Mice. Front. Immunol. 2019, 10, 1424. [Google Scholar] [CrossRef]
- Chen, T.; Zhu, S.; Wei, N.; Zhao, Z.; Niu, J.; Si, Y.; Cao, S.; Ye, J. Protective Immune Responses Induced by an MRNA-LNP Vaccine Encoding PrM-E Proteins against Japanese Encephalitis Virus Infection. Viruses 2022, 14, 1121. [Google Scholar] [CrossRef] [PubMed]
- Medina-Magües, L.G.; Mühe, J.; Jasny, E.; Medina-Magües, E.S.; Roth, N.; Lopera-Madrid, J.; Salas-Quinchucua, C.; Knuese, C.; Petsch, B.; Osorio, J.E. Immunogenicity and Protective Activity of MRNA Vaccine Candidates against Yellow Fever Virus in Animal Models. Npj Vaccines 2023, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 24 June 2023).
- Available online: https://www.cdc.gov/flu/professionals/vaccination/vax-summary.htm (accessed on 25 June 2023).
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. MRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- Available online: https://investors.modernatx.com/news/news-details/2021/Moderna-Announces-Positive-Interim-Phase-1-Data-for-mRNA-Flu-Vaccine-and-Provides-Program-Update/default.aspx (accessed on 25 June 2023).
- Available online: https://s28.q4cdn.com/781576035/files/doc_financials/2022/q2/q2-2022-earnings-charts-final.pdf (accessed on 16 October 2022).
- Available online: https://www.cdc.gov/flu/vaccines-work/vaccineeffect.htm (accessed on 25 June 2023).
- Available online: https://s29.q4cdn.com/435878511/files/doc_downloads/program_detail/2023/05/flu-05-04-23.pdf (accessed on 25 June 2023).
- Schmidt, C.; Schnierle, B.S. Self-Amplifying RNA Vaccine Candidates: Alternative Platforms for MRNA Vaccine Development. Pathogens 2023, 12, 138. [Google Scholar] [CrossRef]
- Arevalo, C.P.; Bolton, M.J.; Le Sage, V.; Ye, N.; Furey, C.; Muramatsu, H.; Alameh, M.-G.; Pardi, N.; Drapeau, E.M.; Parkhouse, K. A Multivalent Nucleoside-Modified MRNA Vaccine against All Known Influenza Virus Subtypes. Science 2022, 378, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Freyn, A.W.; da Silva, J.R.; Rosado, V.C.; Bliss, C.M.; Pine, M.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; de Souza Ferreira, L.C.; Weissman, D. A Multi-Targeting, Nucleoside-Modified MRNA Influenza Virus Vaccine Provides Broad Protection in Mice. Mol. Ther. 2020, 28, 1569–1584. [Google Scholar] [CrossRef]
- McMahon, M.; O’Dell, G.; Tan, J.; Sárközy, A.; Vadovics, M.; Carreño, J.M.; Puente-Massaguer, E.; Muramatsu, H.; Bajusz, C.; Rijnink, W. Assessment of a Quadrivalent Nucleoside-Modified MRNA Vaccine That Protects against Group 2 Influenza Viruses. Proc. Natl. Acad. Sci. USA 2022, 119, e2206333119. [Google Scholar] [CrossRef]
- Mazur, N.I.; Martinón-Torres, F.; Baraldi, E.; Fauroux, B.; Greenough, A.; Heikkinen, T.; Manzoni, P.; Mejias, A.; Nair, H.; Papadopoulos, N.G. Lower Respiratory Tract Infection Caused by Respiratory Syncytial Virus: Current Management and New Therapeutics. Lancet Respir. Med. 2015, 3, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Scheltema, N.M.; Gentile, A.; Lucion, F.; Nokes, D.J.; Munywoki, P.K.; Madhi, S.A.; Groome, M.J.; Cohen, C.; Moyes, J.; Thorburn, K. Global Respiratory Syncytial Virus-Associated Mortality in Young Children (RSV GOLD): A Retrospective Case Series. Lancet Glob. Health 2017, 5, e984–e991. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C. Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Young Children in 2015: A Systematic Review and Modelling Study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D. The Burden of Respiratory Syncytial Virus Infection in Young Children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef]
- Graham, B.S.; Modjarrad, K.; McLellan, J.S. Novel Antigens for RSV Vaccines. Curr. Opin. Immunol. 2015, 35, 30–38. [Google Scholar] [CrossRef]
- Fuentes, S.; Coyle, E.M.; Beeler, J.; Golding, H.; Khurana, S. Antigenic Fingerprinting Following Primary RSV Infection in Young Children Identifies Novel Antigenic Sites and Reveals Unlinked Evolution of Human Antibody Repertoires to Fusion and Attachment Glycoproteins. PLoS Pathog. 2016, 12, e1005554. [Google Scholar] [CrossRef] [PubMed]
- Crank, M.C.; Ruckwardt, T.J.; Chen, M.; Morabito, K.M.; Phung, E.; Costner, P.J.; Holman, L.A.; Hickman, S.P.; Berkowitz, N.M.; Gordon, I.J. A Proof of Concept for Structure-Based Vaccine Design Targeting RSV in Humans. Science 2019, 365, 505–509. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T. Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, C.; Papenburg, J. Rapid Antigen-Based Testing for Respiratory Syncytial Virus: Moving Diagnostics from Bench to Bedside? Future Microbiol. 2013, 8, 435–444. [Google Scholar] [CrossRef]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.-Y. Prefusion F–Specific Antibodies Determine the Magnitude of RSV Neutralizing Activity in Human Sera. Sci. Transl. Med. 2015, 7, 309ra162. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Kim, A.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structural Basis of Respiratory Syncytial Virus Neutralization by Motavizumab. Nat. Struct. Mol. Biol. 2010, 17, 248–250. [Google Scholar] [CrossRef]
- Scott, L.J.; Lamb, H.M. Palivizumab. Drugs 1999, 58, 305–311. [Google Scholar] [CrossRef]
- Qiu, X.; Xu, S.; Lu, Y.; Luo, Z.; Yan, Y.; Wang, C.; Ji, J. Development of MRNA Vaccines against Respiratory Syncytial Virus (RSV). Cytokine Growth Factor Rev. 2022, 68, 37–53. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E.; Scott, D.A.; Gurtman, A.; Zareba, A.; Jansen, K.U.; Gruber, W.C.; Dormitzer, P.R.; Swanson, K.A.; Jiang, Q. Phase 1/2 Randomized Study of the Immunogenicity, Safety, and Tolerability of a Respiratory Syncytial Virus Prefusion F Vaccine in Adults with Concomitant Inactivated Influenza Vaccine. J. Infect. Dis. 2022, 225, 2056–2066. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, I.; Davis, M.G.; Steenackers, K.; Essink, B.; Vandermeulen, C.; Forgarty, C.; Andrews, C.P.; Kerwin, E.; David, M.-P.; Fissette, L. Safety and Immunogenicity of a Respiratory Syncytial Virus Prefusion F (RSVPreF3) Candidate Vaccine in Older Adults: Phase 1/2 Randomized Clinical Trial. J. Infect. Dis. 2023, 227, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. MRNA-Lipid Nanoparticle COVID-19 Vaccines: Structure and Stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Ndeupen, S.; Qin, Z.; Jacobsen, S.; Bouteau, A.; Estanbouli, H.; Igyártó, B.Z. The MRNA-LNP Platform’s Lipid Nanoparticle Component Used in Preclinical Vaccine Studies Is Highly Inflammatory. iScience 2021, 24, 103479. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Tsai, S.J.; Atai, N.A.; Cacciottolo, M.; Nice, J.; Salehi, A.; Guo, C.; Sedgwick, A.; Kanagavelu, S.; Gould, S.J. Exosome-Mediated MRNA Delivery in Vivo Is Safe and Can Be Used to Induce SARS-CoV-2 Immunity. J. Biol. Chem. 2021, 297, 101266. [Google Scholar] [CrossRef]
- Hung, M.E.; Leonard, J.N. A Platform for Actively Loading Cargo RNA to Elucidate Limiting Steps in EV-Mediated Delivery. J. Extracell. Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef]
- Zickler, A.M.; Liang, X.; De Luca, M.; Gupta, D.; Corso, G.; Errichelli, L.; Hean, J.; Kamei, N.; Niu, Z.; Zhou, G.; et al. Novel Endogenous Engineering Platform for Robust Loading and Delivery of Functional MRNA by Extracellular Vesicles; Bioengineering. bioRxiv 2023. [Google Scholar] [CrossRef]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and Origins of Bacterial Membrane Vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Yue, Y.; Zhang, K.; Cheng, K.; Feng, Q.; Ma, N.; Liang, J.; Zhang, T.; Zhang, L.; et al. Rapid Surface Display of MRNA Antigens by Bacteria-Derived Outer Membrane Vesicles for a Personalized Tumor Vaccine. Adv. Mater. 2022, 34, 2109984. [Google Scholar] [CrossRef] [PubMed]
- Jarzebska, N.T.; Mellett, M.; Frei, J.; Kündig, T.M.; Pascolo, S. Protamine-Based Strategies for RNA Transfection. Pharmaceutics 2021, 13, 877. [Google Scholar] [CrossRef] [PubMed]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-Based Vaccines With Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Talebian, S.; Mendes, B.B.; Wallace, G.; Langer, R.; Conde, J.; Shi, J. Hydrogels for RNA Delivery. Nat. Mater. 2023, 22, 818–831. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Name | Developer | Vaccine Type | Participants | Clinical Trial Status | Year of Approval |
|---|---|---|---|---|---|
| Beyfortus (Nirsevimab) | AstraZeneca/Sanofi | Anti-preF mAb (Immune-prophylaxis) | Infants (0 Days to 12 Months) | Phase 3 (NCT03979313): Attended RSV-LRTI through 150 days after the injection: 74.5% | 2022 |
| mRNA-1345 | Moderna | RSV preF (Nucleic acid) | Elderly | Phase 3 (NCT05127434): RSV-LRTD with ≥2 symptoms: 83.7%; RSV-LRTD with ≥3 symptoms: 82.4% | |
| mRNA-1345 | Moderna | RSV preF (Nucleic acid) | Infants (5–24 Months) | Phase 1 (NCT05743881) | |
| Arexvy (RSVPreF3) | GSK | RSV preF (Protein-based particle) | Elderly (≥60 years) | Phase 3 (NCT04886596) (AReSVi 006): RSV-LRTD with ≥1 symptoms: 82.6%; RSV-LRTD with ≥2 symptoms: 94.1% | 2023 |
| Abrysvo (RSV preF) | Pfizer | RSV preF A and RSV preF B (Protein-based particle) | Elderly (60–80 years old) | Phase 3 (NCT05035212): RSV-LRTD with ≥2 symptoms: 66.7%; RSV-LRTD with ≥3 symptoms: 85.7% | 2023 |
| Abrysvo (RSV preF) | Pfizer | RSV preF A and RSV preF B (Protein-based particle) | Maternal Immunization | Phase 3 (NCT04424316): 90-day-old infants with severe MA-LRTI: 81.8% 180-day-old infants with severe MA-LRTI: 69.4% | |
| BARS13 | Advaccine | G protein (Protein-based particle) | Elderly (60–80 years old) | Phase 2 (NCT04681833) | |
| MVA-BN RSV | Bavarian Nordic | F, N, M2-1, G (subtype A), G (subtype B) (Recombinant vector) | Elderly (≥60 years) | Phase 3 (NCT05238025) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, W.; Jiang, L.; Liao, S.; Wu, F.; Yang, G.; Hou, L.; Liu, L.; Pan, X.; Jia, W.; Zhang, Y. Vaccines’ New Era-RNA Vaccine. Viruses 2023, 15, 1760. https://doi.org/10.3390/v15081760
Zhou W, Jiang L, Liao S, Wu F, Yang G, Hou L, Liu L, Pan X, Jia W, Zhang Y. Vaccines’ New Era-RNA Vaccine. Viruses. 2023; 15(8):1760. https://doi.org/10.3390/v15081760
Chicago/Turabian StyleZhou, Wenshuo, Linglei Jiang, Shimiao Liao, Feifei Wu, Guohuan Yang, Li Hou, Lan Liu, Xinping Pan, William Jia, and Yuntao Zhang. 2023. "Vaccines’ New Era-RNA Vaccine" Viruses 15, no. 8: 1760. https://doi.org/10.3390/v15081760