

The Contribution of Microglia and Brain-Infiltrating Macrophages to the Pathogenesis of Neuroinflammatory and Neurodegenerative Diseases during TMEV Infection of the Central Nervous System


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
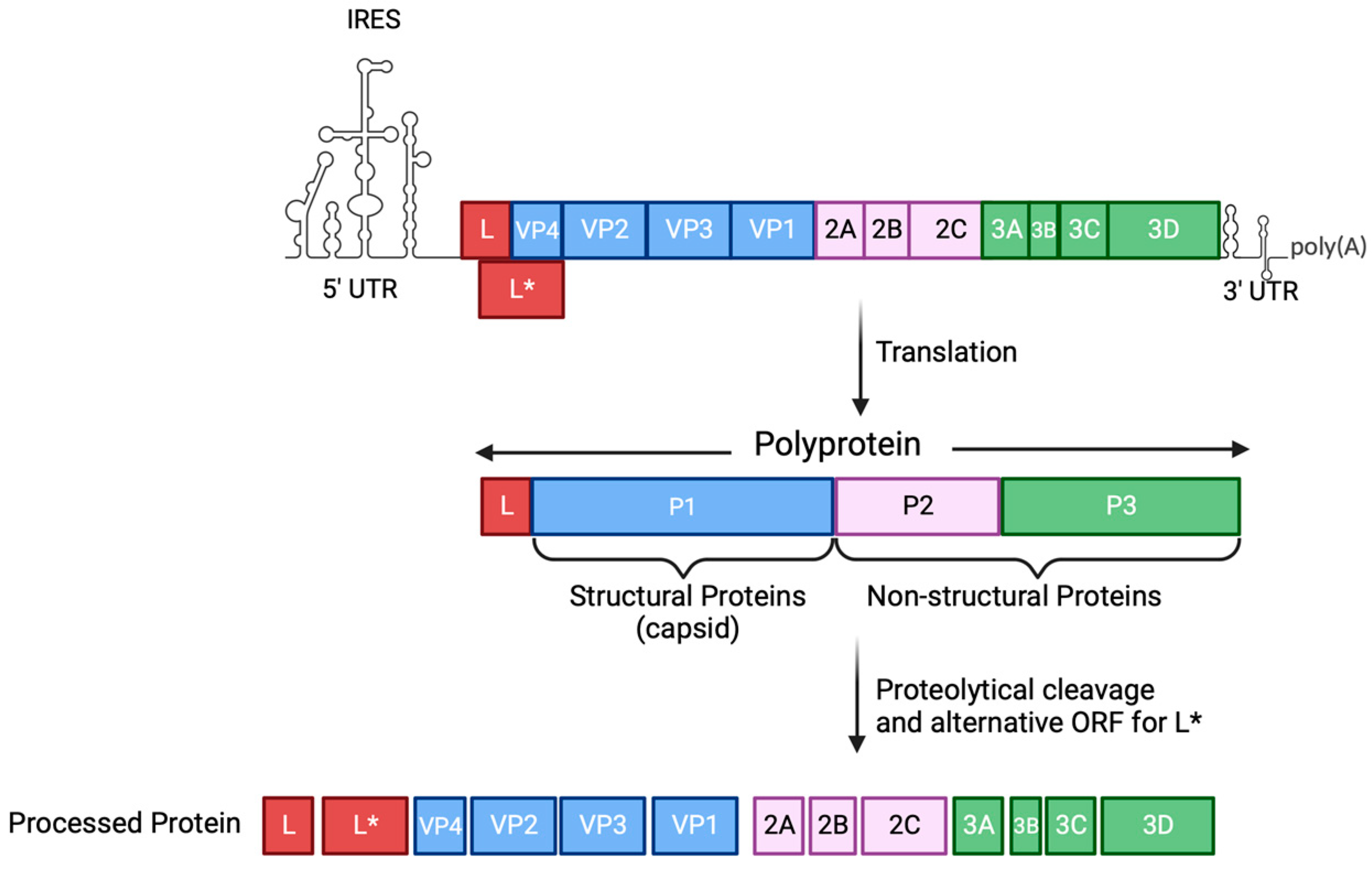
2. An Overview of TMEV
3. TMEV Infection
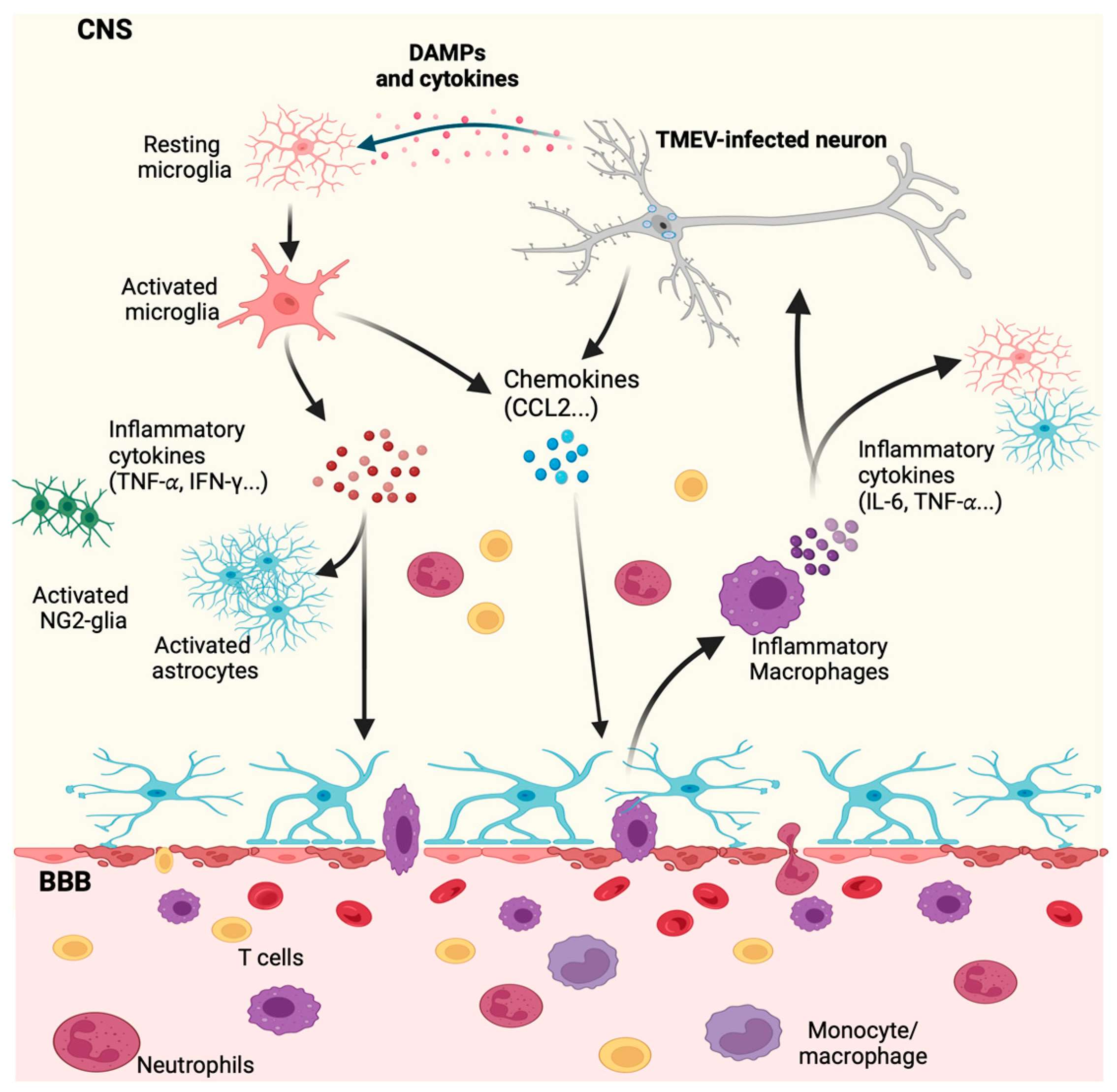
4. Innate Immune Response by Microglia and Macrophages during CNS Viral Infection
4.1. The Role of INF-I Response to TMEV Infection
4.2. TMEV Counteraction of the Interferon Response
5. Multiple Sclerosis
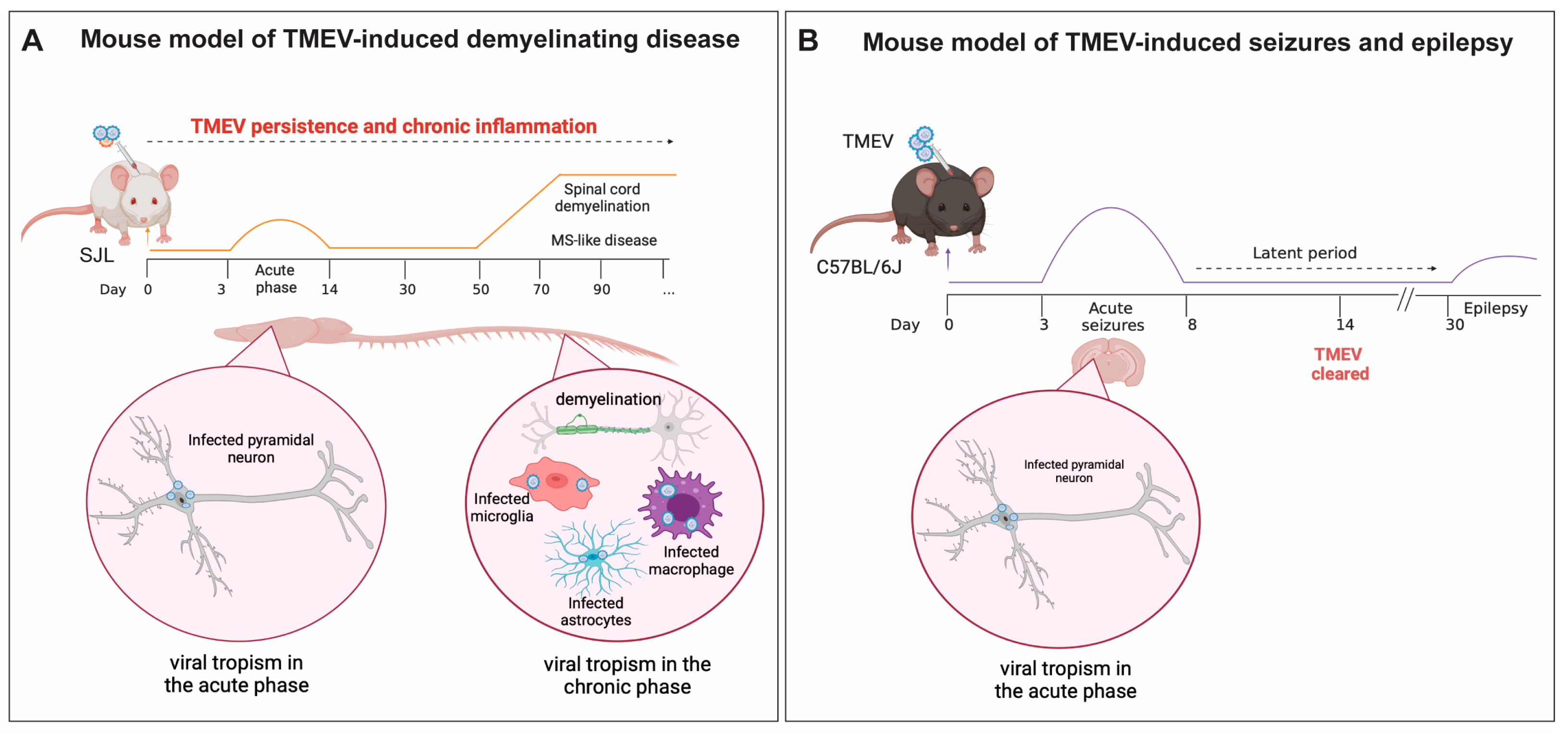
5.1. TMEV-IDD, a Model of Viral-Induced Progressive MS
5.2. The Role of Microglia and Infiltrating Macrophages in Demyelination during TMEV Neurotropic Infection
6. TMEV Infection in a Mouse Model of Viral-Induced Seizures and Epilepsy
The Role of Microglia and Infiltrating Macrophages in Neuronal Excitation Following TMEV CNS Infection
7. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.M.; Sarmast, S.T.; Jahan, N. Viral Infections of the Central Nervous System in Children: A Systematic Review. Cureus 2020, 12, e11174. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Kirkegaard, K.; Macadam, A.; Racaniello, V.R.; Rosenfeld, A.B. The Picornaviridae Family: Knowledge Gaps, Animal Models, Countermeasures, and Prototype Pathogens. J. Infect. Dis. 2023, 228, S427–S445. [Google Scholar] [CrossRef] [PubMed]
- Jubelt, B.; Lipton, H.L. Enterovirus/picornavirus infections. Handb. Clin. Neurol. 2014, 123, 379–416. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.A. Emergent Viral Infections of the CNS. J. Neuropathol. Exp. Neurol. 2020, 79, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Munz, C.; Cohen, J.I.; Ascherio, A. Epstein-Barr virus as a leading cause of multiple sclerosis: Mechanisms and implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef]
- Donati, D. Viral infections and multiple sclerosis. Drug Discov. Today Dis. Models 2020, 32, 27–33. [Google Scholar] [CrossRef]
- Landry, R.L.; Embers, M.E. The Probable Infectious Origin of Multiple Sclerosis. NeuroSci 2023, 4, 211–234. [Google Scholar] [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093.E2. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Jiang, X.; Li, H. The viral hypothesis in Alzheimer’s disease: SARS-CoV-2 on the cusp. Front. Aging Neurosci. 2023, 15, 1129640. [Google Scholar] [CrossRef] [PubMed]
- Sait, A.; Angeli, C.; Doig, A.J.; Day, P.J.R. Viral Involvement in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, L.; Theodore, W.H.; Jacobson, S.; Gaillard, W.D. Infection with HHV-6 and its role in epilepsy. Epilepsy Res. 2019, 153, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Bonello, M.; Michael, B.D.; Solomon, T. Infective Causes of Epilepsy. Semin. Neurol. 2015, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- DePaula-Silva, A.B.; Bell, L.A.; Wallis, G.J.; Wilcox, K.S. Inflammation Unleashed in Viral-Induced Epileptogenesis. Epilepsy Curr. 2021, 21, 433–440. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Loscher, W.; Howe, C.L. Molecular Mechanisms in the Genesis of Seizures and Epilepsy Associated With Viral Infection. Front. Mol. Neurosci. 2022, 15, 870868. [Google Scholar] [CrossRef]
- Misra, U.K.; Tan, C.T.; Kalita, J. Viral encephalitis and epilepsy. Epilepsia 2008, 49 (Suppl. 6), 13–18. [Google Scholar] [CrossRef]
- Pike, S.C.; Welsh, N.; Linzey, M.; Gilli, F. Theiler’s virus-induced demyelinating disease as an infectious model of progressive multiple sclerosis. Front. Mol. Neurosci. 2022, 15, 1019799. [Google Scholar] [CrossRef]
- Sellner, J.; Trinka, E. Seizures and epilepsy in herpes simplex virus encephalitis: Current concepts and future directions of pathogenesis and management. J. Neurol. 2012, 259, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Singhi, P. Infectious causes of seizures and epilepsy in the developing world. Dev. Med. Child. Neurol. 2011, 53, 600–609. [Google Scholar] [CrossRef]
- Suzuki, Y.; Toribe, Y.; Mogami, Y.; Yanagihara, K.; Nishikawa, M. Epilepsy in patients with congenital cytomegalovirus infection. Brain Dev. 2008, 30, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Wilson, S.E.; Rubinos, C. SARS-CoV-2 infection and seizures: The perfect storm. J. Integr. Neurosci. 2022, 21, 115. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.M.; Blumcke, I.; Sander, J.W.; Loscher, W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Bohmwald, K.; Galvez, N.M.S.; Rios, M.; Kalergis, A.M. Neurologic Alterations Due to Respiratory Virus Infections. Front. Cell Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef]
- Hopkins, H.K.; Traverse, E.M.; Barr, K.L. Viral Parkinsonism: An underdiagnosed neurological complication of Dengue virus infection. PLoS Negl. Trop. Dis. 2022, 16, e0010118. [Google Scholar] [CrossRef]
- van den Pol, A.N. Viral infection leading to brain dysfunction: More prevalent than appreciated? Neuron 2009, 64, 17–20. [Google Scholar] [CrossRef]
- Wouk, J.; Rechenchoski, D.Z.; Rodrigues, B.C.D.; Ribelato, E.V.; Faccin-Galhardi, L.C. Viral infections and their relationship to neurological disorders. Arch. Virol. 2021, 166, 733–753. [Google Scholar] [CrossRef]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front. Mol. Neurosci. 2018, 11, 63. [Google Scholar] [CrossRef]
- Carocci, M.; Bakkali-Kassimi, L. The encephalomyocarditis virus. Virulence 2012, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Erkens Hulshof, S.; Lanke, K.; Verduyn Lunel, F.; Melchers, W.J.; Schoondermark-van de Ven, E.; Roivainen, M.; Galama, J.M.; van Kuppeveld, F.J. Saffold virus, a human Theiler’s-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 2009, 5, e1000416. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Fujinami, R.S. Neurotropic viral infections leading to epilepsy: Focus on Theiler’s murine encephalomyelitis virus. Future Virol. 2011, 6, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Kim, B.S.; Yahikozawa, H.; Nadler, C.F. Serological evidence that Mus musculus is the natural host of Theiler’s murine encephalomyelitis virus. Virus Res. 2001, 76, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Aubert, C.; Chamorro, M.; Brahic, M. Identification of Theiler’s virus infected cells in the central nervous system of the mouse during demyelinating disease. Microb. Pathog. 1987, 3, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L. Theiler’s virus infection in mice: An unusual biphasic disease process leading to demyelination. Infect. Immun. 1975, 11, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Theiler, M. Spontaneous Encephalomyelitis of Mice, a New Virus Disease. J. Exp. Med. 1937, 65, 705–719. [Google Scholar] [CrossRef]
- Daniels, J.B.; Pappenheimer, A.M.; Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 1952, 96, 517–530. [Google Scholar] [CrossRef]
- DePaula-Silva, A.B.; Hanak, T.J.; Libbey, J.E.; Fujinami, R.S. Theiler’s murine encephalomyelitis virus infection of SJL/J and C57BL/6J mice: Models for multiple sclerosis and epilepsy. J. Neuroimmunol. 2017, 308, 30–42. [Google Scholar] [CrossRef]
- Gerhauser, I.; Hansmann, F.; Ciurkiewicz, M.; Loscher, W.; Beineke, A. Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update. Int. J. Mol. Sci. 2019, 20, 448. [Google Scholar] [CrossRef]
- Jarousse, N.; Syan, S.; Martinat, C.; Brahic, M. The neurovirulence of the DA and GDVII strains of Theiler’s virus correlates with their ability To infect cultured neurons. J. Virol. 1998, 72, 7213–7220. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S. Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler’s Virus-Induced Demyelinating Disease. Int. J. Mol. Sci. 2021, 22, 5254. [Google Scholar] [CrossRef]
- Lipton, H.L.; Jelachich, M.L. Molecular pathogenesis of Theiler’s murine encephalomyelitis virus-induced demyelinating disease in mice. Intervirology 1997, 40, 143–152. [Google Scholar]
- Broer, S.; Hage, E.; Kaufer, C.; Gerhauser, I.; Anjum, M.; Li, L.; Baumgartner, W.; Schulz, T.F.; Loscher, W. Viral mouse models of multiple sclerosis and epilepsy: Marked differences in neuropathogenesis following infection with two naturally occurring variants of Theiler’s virus BeAn strain. Neurobiol. Dis. 2017, 99, 121–132. [Google Scholar] [CrossRef]
- Buhler, M.; Runft, S.; Li, D.; Gotting, J.; Detje, C.N.; Nippold, V.; Stoff, M.; Beineke, A.; Schulz, T.; Kalinke, U.; et al. IFN-beta Deficiency Results in Fatal or Demyelinating Disease in C57BL/6 Mice Infected with Theiler’s Murine Encephalomyelitis Viruses. Front. Immunol. 2022, 13, 786940. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Kirkman, N.J.; Smith, M.C.; Tanaka, T.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Seizures following picornavirus infection. Epilepsia 2008, 49, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Fujinami, R.S. Viral mouse models used to study multiple sclerosis: Past and present. Arch. Virol. 2021, 166, 1015–1033. [Google Scholar] [CrossRef]
- Omura, S.; Kawai, E.; Sato, F.; Martinez, N.E.; Chaitanya, G.V.; Rollyson, P.A.; Cvek, U.; Trutschl, M.; Alexander, J.S.; Tsunoda, I. Bioinformatics multivariate analysis determined a set of phase-specific biomarker candidates in a novel mouse model for viral myocarditis. Circ. Cardiovasc. Genet. 2014, 7, 444–454. [Google Scholar] [CrossRef]
- Sato, F.; Omura, S.; Kawai, E.; Martinez, N.E.; Acharya, M.M.; Reddy, P.C.; Chaitanya, G.V.; Alexander, J.S.; Tsunoda, I. Distinct kinetics of viral replication, T cell infiltration, and fibrosis in three phases of myocarditis following Theiler’s virus infection. Cell Immunol. 2014, 292, 85–93. [Google Scholar] [CrossRef]
- Tsunoda, I.; Sato, F.; Omura, S.; Fujita, M.; Sakiyama, N.; Park, A.M. Three immune-mediated disease models induced by Theiler’s virus: Multiple sclerosis, seizures and myocarditis. Clin. Exp. Neuroimmunol. 2016, 7, 330–345. [Google Scholar] [CrossRef]
- Lipton, H.L.; Kumar, A.S.; Trottier, M. Theiler’s virus persistence in the central nervous system of mice is associated with continuous viral replication and a difference in outcome of infection of infiltrating macrophages versus oligodendrocytes. Virus Res. 2005, 111, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Libbey, J.E.; Fujinami, R.S. Theiler’s murine encephalomyelitis virus attachment to the gastrointestinal tract is associated with sialic acid binding. J. Neurovirol. 2009, 15, 81–89. [Google Scholar] [CrossRef]
- Caliskan, N.; Hill, C.H. Insights from structural studies of the cardiovirus 2A protein. Biosci. Rep. 2022, 42, BSR20210406. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Drappier, M.; Michiels, T. Innate Immune Detection of Cardioviruses and Viral Disruption of Interferon Signaling. Front. Microbiol. 2018, 9, 2448. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Chen, T.C.; Weng, K.F.; Chang, S.C.; Chen, L.L.; Shih, S.R. Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 2009, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Paget, M.; Wang, C.; Zhu, Z.; Zheng, H. Innate immune evasion by picornaviruses. Eur. J. Immunol. 2020, 50, 1268–1282. [Google Scholar] [CrossRef]
- Ghadge, G.D.; Ma, L.; Sato, S.; Kim, J.; Roos, R.P. A protein critical for a Theiler’s virus-induced immune system-mediated demyelinating disease has a cell type-specific antiapoptotic effect and a key role in virus persistence. J. Virol. 1998, 72, 8605–8612. [Google Scholar] [CrossRef]
- Ohara, Y.; Stein, S.; Fu, J.L.; Stillman, L.; Klaman, L.; Roos, R.P. Molecular cloning and sequence determination of DA strain of Theiler’s murine encephalomyelitis viruses. Virology 1988, 164, 245–255. [Google Scholar] [CrossRef]
- Bell, L.A.; Wallis, G.J.; Wilcox, K.S. Reactivity and increased proliferation of NG2 cells following central nervous system infection with Theiler’s murine encephalomyelitis virus. J. Neuroinflamm. 2020, 17, 369. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gomez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Waltl, I.; Kaufer, C.; Broer, S.; Chhatbar, C.; Ghita, L.; Gerhauser, I.; Anjum, M.; Kalinke, U.; Loscher, W. Macrophage depletion by liposome-encapsulated clodronate suppresses seizures but not hippocampal damage after acute viral encephalitis. Neurobiol. Dis. 2018, 110, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. The innate immune response affects the development of the autoimmune response in Theiler’s virus-induced demyelinating disease. J. Immunol. 2009, 182, 5712–5722. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; LaFrance-Corey, R.G.; Overlee, B.L.; Johnson, R.K.; Clarkson, B.D.S.; Goddery, E.N. Inflammatory monocytes and microglia play independent roles in inflammatory ictogenesis. J. Neuroinflamm. 2022, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Cusick, M.F.; Libbey, J.E.; Patel, D.C.; Doty, D.J.; Fujinami, R.S. Infiltrating macrophages are key to the development of seizures following virus infection. J. Virol. 2013, 87, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- DePaula-Silva, A.B.; Sonderegger, F.L.; Libbey, J.E.; Doty, D.J.; Fujinami, R.S. The immune response to picornavirus infection and the effect of immune manipulation on acute seizures. J. Neurovirol. 2018, 24, 464–477. [Google Scholar] [CrossRef]
- Patel, D.C.; Wallis, G.; Dahle, E.J.; McElroy, P.B.; Thomson, K.E.; Tesi, R.J.; Szymkowski, D.E.; West, P.J.; Smeal, R.M.; Patel, M.; et al. Hippocampal TNFalpha Signaling Contributes to Seizure Generation in an Infection-Induced Mouse Model of Limbic Epilepsy. eNeuro 2017, 4, 1–20. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Lu, H.; Miranda, A.S.; Ransohoff, R.M. Ontogeny and functions of central nervous system macrophages. J. Immunol. 2014, 193, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.E6. [Google Scholar] [CrossRef]
- Bosco, D.B.; Tian, D.S.; Wu, L.J. Neuroimmune interaction in seizures and epilepsy: Focusing on monocyte infiltration. FEBS J. 2020, 287, 4822–4837. [Google Scholar] [CrossRef]
- Filgueira, L.; Larionov, A.; Lannes, N. The Influence of Virus Infection on Microglia and Accelerated Brain Aging. Cells 2021, 10, 1836. [Google Scholar] [CrossRef]
- Chhatbar, C.; Prinz, M. The roles of microglia in viral encephalitis: From sensome to therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 250–258. [Google Scholar] [CrossRef]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Coutinho-Silva, R. Purinergic signaling, DAMPs, and inflammation. Am. J. Physiol. Cell Physiol. 2020, 318, C832–C835. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Waltl, I.; Kalinke, U. Beneficial and detrimental functions of microglia during viral encephalitis. Trends Neurosci. 2022, 45, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; LaFrance-Corey, R.G.; Goddery, E.N.; Johnson, R.K.; Mirchia, K. Neuronal CCL2 expression drives inflammatory monocyte infiltration into the brain during acute virus infection. J. Neuroinflamm. 2017, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ganguly, A.; Zhao, J.; Ivey, M.; Lopez, R.; Osterholzer, J.J.; Cho, C.S.; Olszewski, M.A. CCR2 Signaling Promotes Brain Infiltration of Inflammatory Monocytes and Contributes to Neuropathology during Cryptococcal Meningoencephalitis. mBio 2021, 12, e0107621. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Blank, A.; Bungert, A.D.; Vajkoczy, P. Distinction of Microglia and Macrophages in Glioblastoma: Close Relatives, Different Tasks? Int. J. Mol. Sci. 2020, 22, 194. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- DePaula-Silva, A.B.; Gorbea, C.; Doty, D.J.; Libbey, J.E.; Sanchez, J.M.S.; Hanak, T.J.; Cazalla, D.; Fujinami, R.S. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflamm. 2019, 16, 152. [Google Scholar] [CrossRef]
- Mercurio, D.; Fumagalli, S.; Schafer, M.K.; Pedragosa, J.; Ngassam, L.D.C.; Wilhelmi, V.; Winterberg, S.; Planas, A.M.; Weihe, E.; De Simoni, M.G. Protein Expression of the Microglial Marker Tmem119 Decreases in Association With Morphological Changes and Location in a Mouse Model of Traumatic Brain Injury. Front. Cell Neurosci. 2022, 16, 820127. [Google Scholar] [CrossRef]
- Vankriekelsvenne, E.; Chrzanowski, U.; Manzhula, K.; Greiner, T.; Wree, A.; Hawlitschka, A.; Llovera, G.; Zhan, J.; Joost, S.; Schmitz, C.; et al. Transmembrane protein 119 is neither a specific nor a reliable marker for microglia. Glia 2022, 70, 1170–1190. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Tang, T.M.; Mendsaikhan, A.; Tooyama, I.; Serrano, G.E.; Sue, L.I.; Beach, T.G.; Lue, L.F. Patterns of Expression of Purinergic Receptor P2RY12, a Putative Marker for Non-Activated Microglia, in Aged and Alzheimer’s Disease Brains. Int. J. Mol. Sci. 2020, 21, 678. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabuddhe, V.; Ghosh, H.S. Cx3Cr1-Cre induction leads to microglial activation and IFN-1 signaling caused by DNA damage in early postnatal brain. Cell Rep. 2022, 38, 110252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.F.; Alam, M.M.; Liao, Y.; Huang, T.; Mathur, R.; Zhu, X.; Huang, Y. Targeting Microglia Using Cx3cr1-Cre Lines: Revisiting the Specificity. eNeuro 2019, 6, 1–11. [Google Scholar] [CrossRef]
- Komiya, H.; Takeuchi, H.; Ogawa, Y.; Hatooka, Y.; Takahashi, K.; Katsumoto, A.; Kubota, S.; Nakamura, H.; Kunii, M.; Tada, M.; et al. CCR2 is localized in microglia and neurons, as well as infiltrating monocytes, in the lumbar spinal cord of ALS mice. Mol. Brain 2020, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A. Interferons alpha and beta as immune regulators—A new look. Immunity 2001, 14, 661–664. [Google Scholar] [CrossRef]
- Li, Y.; Yu, P.; Qu, C.; Li, P.; Li, Y.; Ma, Z.; Wang, W.; de Man, R.A.; Peppelenbosch, M.P.; Pan, Q. MDA5 against enteric viruses through induction of interferon-like response partially via the JAK-STAT cascade. Antivir. Res. 2020, 176, 104743. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017, 69, 297–304. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, S.; Anfossi, R.; Humeres, C.; Vivar, R.; Boza, P.; Munoz, C.; Pardo-Jimenez, V.; Olivares-Silva, F.; Diaz-Araya, G. IFN-beta Plays Both Pro- and Anti-inflammatory Roles in the Rat Cardiac Fibroblast Through Differential STAT Protein Activation. Front. Pharmacol. 2018, 9, 1368. [Google Scholar] [CrossRef]
- Song, J.; Guan, M.; Zhao, Z.; Zhang, J. Type I Interferons Function as Autocrine and Paracrine Factors to Induce Autotaxin in Response to TLR Activation. PLoS ONE 2015, 10, e0136629. [Google Scholar] [CrossRef]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Karpus, W.J.; Kennedy, K.J.; Fife, B.T.; Bennett, J.L.; Dal Canto, M.C.; Kunkel, S.L.; Lukacs, N.W. Anti-CCL2 treatment inhibits Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neurovirol. 2006, 12, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Paty, D.W.; Kim, S.U. Differential effects of beta and gamma interferons on expression of major histocompatibility complex antigens and intercellular adhesion molecule-1 in cultured fetal human astrocytes. Neurology 1995, 45, 367–373. [Google Scholar] [CrossRef]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 2014, 5, e01476-01414. [Google Scholar] [CrossRef]
- Stone, L.A.; Frank, J.A.; Albert, P.S.; Bash, C.; Smith, M.E.; Maloni, H.; McFarland, H.F. The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann. Neurol. 1995, 37, 611–619. [Google Scholar] [CrossRef]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Matsumiya, T.; Shiba, Y.; Ding, J.; Kawaguchi, S.; Seya, K.; Imaizumi, T. The double-stranded RNA-dependent protein kinase PKR negatively regulates the protein expression of IFN-beta induced by RIG-I signaling. FASEB J. 2023, 37, e22780. [Google Scholar] [CrossRef]
- Kang, M.H.; So, E.Y.; Park, H.; Kim, B.S. Replication of Theiler’s virus requires NF-kappa B-activation: Higher viral replication and spreading in astrocytes from susceptible mice. Glia 2008, 56, 942–953. [Google Scholar] [CrossRef]
- Drappier, M.; Jha, B.K.; Stone, S.; Elliott, R.; Zhang, R.; Vertommen, D.; Weiss, S.R.; Silverman, R.H.; Michiels, T. A novel mechanism of RNase L inhibition: Theiler’s virus L* protein prevents 2-5A from binding to RNase L. PLoS Pathog. 2018, 14, e1006989. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Maroney, P.A.; Baglioni, C. Double-stranded RNA causes synthesis of 2’,5’-oligo(A) and degradation of messenger RNA in interferon-treated cells. J. Biol. Chem. 1981, 256, 7806–7811. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C.; et al. Interferon action and apoptosis are defective in mice devoid of 2’,5’-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Ricour, C.; Delhaye, S.; Hato, S.V.; Olenyik, T.D.; Michel, B.; van Kuppeveld, F.J.; Gustin, K.E.; Michiels, T. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler’s virus leader protein. J. Gen. Virol. 2009, 90, 177–186. [Google Scholar] [CrossRef]
- Sorgeloos, F.; Jha, B.K.; Silverman, R.H.; Michiels, T. Evasion of antiviral innate immunity by Theiler’s virus L* protein through direct inhibition of RNase L. PLoS Pathog. 2013, 9, e1003474. [Google Scholar] [CrossRef]
- Stavrou, S.; Feng, Z.; Lemon, S.M.; Roos, R.P. Different strains of Theiler’s murine encephalomyelitis virus antagonize different sites in the type I interferon pathway. J. Virol. 2010, 84, 9181–9189. [Google Scholar] [CrossRef]
- Borghese, F.; Michiels, T. The leader protein of cardioviruses inhibits stress granule assembly. J. Virol. 2011, 85, 9614–9622. [Google Scholar] [CrossRef]
- Takano-Maruyama, M.; Ohara, Y.; Asakura, K.; Okuwa, T. Leader (L) and L* proteins of Theiler’s murine encephalomyelitis virus (TMEV) and their regulation of the virus’ biological activities. J. Neuroinflamm. 2006, 3, 19. [Google Scholar] [CrossRef]
- Miyamoto, M.; Himeda, T.; Ishihara, K.; Okuwa, T.; Kobayashi, D.; Nameta, M.; Karasawa, Y.; Chunhaphinyokul, B.; Yoshida, Y.; Tanaka, N.; et al. Theilovirus 3C Protease Cleaves the C-Terminal Domain of the Innate Immune RNA Sensor, Melanoma Differentiation-Associated Gene 5, and Impairs Double-Stranded RNA-Mediated IFN Response. J. Immunol. 2023, 210, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Smith, K.A.; Hiyoshi, A.; Piehl, F.; Olsson, T.; Montgomery, S. Hospital-diagnosed infections before age 20 and risk of a subsequent multiple sclerosis diagnosis. Brain 2021, 144, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Leray, E.; Moreau, T.; Fromont, A.; Edan, G. Epidemiology of multiple sclerosis. Rev. Neurol. 2016, 172, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Bjartmar, C.; Kidd, G.; Mork, S.; Rudick, R.; Trapp, B.D. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann. Neurol. 2000, 48, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Cox, A.; Le Page, E.; Jones, J.; Trip, S.A.; Deans, J.; Seaman, S.; Miller, D.H.; Hale, G.; Waldmann, H.; et al. The window of therapeutic opportunity in multiple sclerosis: Evidence from monoclonal antibody therapy. J. Neurol. 2006, 253, 98–108. [Google Scholar] [CrossRef]
- Karussis, D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: A critical review. J. Autoimmun. 2014, 48–49, 134–142. [Google Scholar] [CrossRef]
- Rodgers, M.M.; Mulcare, J.A.; King, D.L.; Mathews, T.; Gupta, S.C.; Glaser, R.M. Gait characteristics of individuals with multiple sclerosis before and after a 6-month aerobic training program. J. Rehabil. Res. Dev. 1999, 36, 183–188. [Google Scholar]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Mudge, J.; van Velkinburgh, J.C.; Khankhanian, P.; Khrebtukova, I.; Miller, N.A.; Zhang, L.; Farmer, A.D.; Bell, C.J.; Kim, R.W.; et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010, 464, 1351–1356. [Google Scholar] [CrossRef]
- Ingelfinger, F.; Gerdes, L.A.; Kavaka, V.; Krishnarajah, S.; Friebel, E.; Galli, E.; Zwicky, P.; Furrer, R.; Peukert, C.; Dutertre, C.A.; et al. Twin study reveals non-heritable immune perturbations in multiple sclerosis. Nature 2022, 603, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Kakalacheva, K.; Munz, C.; Lunemann, J.D. Viral triggers of multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Swanson, P.A., 2nd; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef]
- Makhani, N.; Banwell, B.; Tellier, R.; Yea, C.; McGovern, S.; O’Mahony, J.; Ahorro, J.M.; Arnold, D.; Sadovnick, A.D.; Marrie, R.A.; et al. Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult. Scler. 2016, 22, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef]
- Stohlman, S.A.; Ramakrishna, C.; Tschen, S.I.; Hinton, D.R.; Bergmann, C.C. The art of survival during viral persistence. J. Neurovirol. 2002, 8 (Suppl. 2), 53–58. [Google Scholar] [CrossRef]
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643. [Google Scholar] [CrossRef]
- Ghaderi, S.; Berg-Hansen, P.; Bakken, I.J.; Magnus, P.; Trogstad, L.; Haberg, S.E. Hospitalization following influenza infection and pandemic vaccination in multiple sclerosis patients: A nationwide population-based registry study from Norway. Eur. J. Epidemiol. 2020, 35, 355–362. [Google Scholar] [CrossRef]
- Steelman, A.J. Infection as an Environmental Trigger of Multiple Sclerosis Disease Exacerbation. Front. Immunol. 2015, 6, 520. [Google Scholar] [CrossRef] [PubMed]
- Clatch, R.J.; Miller, S.D.; Metzner, R.; Dal Canto, M.C.; Lipton, H.L. Monocytes/macrophages isolated from the mouse central nervous system contain infectious Theiler’s murine encephalomyelitis virus (TMEV). Virology 1990, 176, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Trottier, M.; Kallio, P.; Wang, W.; Lipton, H.L. High numbers of viral RNA copies in the central nervous system of mice during persistent infection with Theiler’s virus. J. Virol. 2001, 75, 7420–7428. [Google Scholar] [CrossRef] [PubMed]
- DiSano, K.D.; Linzey, M.R.; Royce, D.B.; Pachner, A.R.; Gilli, F. Differential neuro-immune patterns in two clinically relevant murine models of multiple sclerosis. J. Neuroinflamm. 2019, 16, 109. [Google Scholar] [CrossRef] [PubMed]
- DiSano, K.D.; Royce, D.B.; Gilli, F.; Pachner, A.R. Central Nervous System Inflammatory Aggregates in the Theiler’s Virus Model of Progressive Multiple Sclerosis. Front. Immunol. 2019, 10, 1821. [Google Scholar] [CrossRef]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse models of multiple sclerosis: Experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol. Biol. 2012, 900, 381–401. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Calenoff, M.A.; Miller, S.D.; Vanderlugt, C.L. Lymphocytes from mice chronically infected with Theiler’s murine encephalomyelitis virus produce demyelination of organotypic cultures after stimulation with the major encephalitogenic epitope of myelin proteolipid protein. Epitope spreading in TMEV infection has functional activity. J. Neuroimmunol. 2000, 104, 79–84. [Google Scholar] [CrossRef]
- Neville, K.L.; Padilla, J.; Miller, S.D. Myelin-specific tolerance attenuates the progression of a virus-induced demyelinating disease: Implications for the treatment of MS. J. Neuroimmunol. 2002, 123, 18–29. [Google Scholar] [CrossRef]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central nervous system macrophages in progressive multiple sclerosis: Relationship to neurodegeneration and therapeutics. J. Neuroinflamm. 2022, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Touil, H.; Pikor, N.B.; Gommerman, J.L.; Prat, A.; Bar-Or, A. B Cells in the Multiple Sclerosis Central Nervous System: Trafficking and Contribution to CNS-Compartmentalized Inflammation. Front. Immunol. 2015, 6, 636. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cerda, F.; Sanchez-Gomes, M.V.; Matute, C. The link of inflammation and neurodegeneration in progressive multiple sclerosis. Mult. Scler. Demyelinating Disord. 2016, 1, 9. [Google Scholar] [CrossRef]
- Howe, C.L.; Lafrance-Corey, R.G.; Sundsbak, R.S.; Lafrance, S.J. Inflammatory monocytes damage the hippocampus during acute picornavirus infection of the brain. J. Neuroinflamm. 2012, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Lafrance-Corey, R.G.; Sundsbak, R.S.; Sauer, B.M.; Lafrance, S.J.; Buenz, E.J.; Schmalstieg, W.F. Hippocampal protection in mice with an attenuated inflammatory monocyte response to acute CNS picornavirus infection. Sci. Rep. 2012, 2, 545. [Google Scholar] [CrossRef]
- Buenz, E.J.; Rodriguez, M.; Howe, C.L. Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol. Dis. 2006, 24, 266–273. [Google Scholar] [CrossRef]
- Buenz, E.J.; Sauer, B.M.; Lafrance-Corey, R.G.; Deb, C.; Denic, A.; German, C.L.; Howe, C.L. Apoptosis of hippocampal pyramidal neurons is virus independent in a mouse model of acute neurovirulent picornavirus infection. Am. J. Pathol. 2009, 175, 668–684. [Google Scholar] [CrossRef]
- Uhde, A.K.; Ciurkiewicz, M.; Herder, V.; Khan, M.A.; Hensel, N.; Claus, P.; Beckstette, M.; Teich, R.; Floess, S.; Baumgartner, W.; et al. Intact interleukin-10 receptor signaling protects from hippocampal damage elicited by experimental neurotropic virus infection of SJL mice. Sci. Rep. 2018, 8, 6106. [Google Scholar] [CrossRef]
- Kummerfeld, M.; Seehusen, F.; Klein, S.; Ulrich, R.; Kreutzer, R.; Gerhauser, I.; Herder, V.; Baumgartner, W.; Beineke, A. Periventricular demyelination and axonal pathology is associated with subependymal virus spread in a murine model for multiple sclerosis. Intervirology 2012, 55, 401–416. [Google Scholar] [CrossRef]
- Martinat, C.; Jarousse, N.; Prevost, M.C.; Brahic, M. The GDVII strain of Theiler’s virus spreads via axonal transport. J. Virol. 1999, 73, 6093–6098. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Aubert, C.; Brahic, M. Theiler’s virus replication in brain macrophages cultured in vitro. J. Virol. 1992, 66, 3188–3193. [Google Scholar] [CrossRef]
- Rodriguez, M.; Leibowitz, J.L.; Lampert, P.W. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Ann. Neurol. 1983, 13, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Twaddle, G.; Jelachich, M.L. The predominant virus antigen burden is present in macrophages in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Virol. 1995, 69, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Calenoff, M.A.; Dal Canto, M.C. Astrocytes, not microglia, are the main cells responsible for viral persistence in Theiler’s murine encephalomyelitis virus infection leading to demyelination. J. Neuroimmunol. 2001, 118, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef]
- Olson, J.K.; Croxford, J.L.; Calenoff, M.A.; Dal Canto, M.C.; Miller, S.D. A virus-induced molecular mimicry model of multiple sclerosis. J. Clin. Investig. 2001, 108, 311–318. [Google Scholar] [CrossRef]
- Olson, J.K.; Ercolini, A.M.; Miller, S.D. A virus-induced molecular mimicry model of multiple sclerosis. Curr. Top. Microbiol. Immunol. 2005, 296, 39–53. [Google Scholar] [CrossRef]
- Ciurkiewicz, M.; Floess, S.; Beckstette, M.; Kummerfeld, M.; Baumgartner, W.; Huehn, J.; Beineke, A. Transcriptome analysis following neurotropic virus infection reveals faulty innate immunity and delayed antigen presentation in mice susceptible to virus-induced demyelination. Brain Pathol. 2021, 31, e13000. [Google Scholar] [CrossRef]
- Kim, B.S. Critical role of TLR activation in viral replication, persistence, and pathogenicity of Theiler’s virus. Front. Immunol. 2023, 14, 1167972. [Google Scholar] [CrossRef]
- Bowen, J.L.; Olson, J.K. Innate immune CD11b+Gr-1+ cells, suppressor cells, affect the immune response during Theiler’s virus-induced demyelinating disease. J. Immunol. 2009, 183, 6971–6980. [Google Scholar] [CrossRef] [PubMed]
- Stohlman, S.A.; Hinton, D.R. Viral induced demyelination. Brain Pathol. 2001, 11, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Lipton, H.L. Ultrastructural immunohistochemical localization of virus in acute and chronic demyelinating Theiler’s virus infection. Am. J. Pathol. 1982, 106, 20–29. [Google Scholar] [PubMed]
- Rossi, C.P.; Delcroix, M.; Huitinga, I.; McAllister, A.; van Rooijen, N.; Claassen, E.; Brahic, M. Role of macrophages during Theiler’s virus infection. J. Virol. 1997, 71, 3336–3340. [Google Scholar] [CrossRef] [PubMed]
- Luong, N.; Olson, J.K. Exosomes Secreted by Microglia During Virus Infection in the Central Nervous System Activate an Inflammatory Response in Bystander Cells. Front. Cell Dev. Biol. 2021, 9, 661935. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Girvin, A.M.; Miller, S.D. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J. Virol. 2001, 75, 9780–9789. [Google Scholar] [CrossRef]
- Jin, Y.H.; Kim, S.J.; So, E.Y.; Meng, L.; Colonna, M.; Kim, B.S. Melanoma differentiation-associated gene 5 is critical for protection against Theiler’s virus-induced demyelinating disease. J. Virol. 2012, 86, 1531–1543. [Google Scholar] [CrossRef]
- Goddery, E.N.; Fain, C.E.; Lipovsky, C.G.; Ayasoufi, K.; Yokanovich, L.T.; Malo, C.S.; Khadka, R.H.; Tritz, Z.P.; Jin, F.; Hansen, M.J.; et al. Microglia and Perivascular Macrophages Act as Antigen Presenting Cells to Promote CD8 T Cell Infiltration of the Brain. Front. Immunol. 2021, 12, 726421. [Google Scholar] [CrossRef]
- Moseman, E.A.; Blanchard, A.C.; Nayak, D.; McGavern, D.B. T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci. Immunol. 2020, 5, eabb1817. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2017, 8, 1905. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Hammel, G.; Zivkovic, S.; Ayazi, M.; Ren, Y. Consequences and mechanisms of myelin debris uptake and processing by cells in the central nervous system. Cell Immunol. 2022, 380, 104591. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Sanchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.E.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Javonillo, D.I.; Pachow, C.; Scarfone, V.M.; Fernandez, K.; Walsh, C.M.; Green, K.N.; Lane, T.E. Ablation of microglia following infection of the central nervous system with a neurotropic murine coronavirus infection leads to increased demyelination and impaired remyelination. J. Neuroimmunol. 2023, 381, 578133. [Google Scholar] [CrossRef]
- Sariol, A.; Mackin, S.; Allred, M.G.; Ma, C.; Zhou, Y.; Zhang, Q.; Zou, X.; Abrahante, J.E.; Meyerholz, D.K.; Perlman, S. Microglia depletion exacerbates demyelination and impairs remyelination in a neurotropic coronavirus infection. Proc. Natl. Acad. Sci. USA 2020, 117, 24464–24474. [Google Scholar] [CrossRef]
- Sanchez, J.M.S.; DePaula-Silva, A.B.; Doty, D.J.; Hanak, T.J.; Truong, A.; Libbey, J.E.; Fujinami, R.S. The CSF1R-Microglia Axis Has Protective Host-Specific Roles During Neurotropic Picornavirus Infection. Front. Immunol. 2021, 12, 621090. [Google Scholar] [CrossRef] [PubMed]
- Waltl, I.; Kaufer, C.; Gerhauser, I.; Chhatbar, C.; Ghita, L.; Kalinke, U.; Loscher, W. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav. Immun. 2018, 74, 186–204. [Google Scholar] [CrossRef]
- Bruck, W.; Porada, P.; Poser, S.; Rieckmann, P.; Hanefeld, F.; Kretzschmar, H.A.; Lassmann, H. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann. Neurol. 1995, 38, 788–796. [Google Scholar] [CrossRef]
- Popescu, B.F.; Lucchinetti, C.F. Pathology of demyelinating diseases. Annu. Rev. Pathol. 2012, 7, 185–217. [Google Scholar] [CrossRef]
- Bruck, W.; Sommermeier, N.; Bergmann, M.; Zettl, U.; Goebel, H.H.; Kretzschmar, H.A.; Lassmann, H. Macrophages in multiple sclerosis. Immunobiology 1996, 195, 588–600. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Fife, B.T.; Begolka, W.S.; Miller, S.D.; Karpus, W.J. Central nervous system chemokine expression during Theiler’s virus-induced demyelinating disease. J. Neurovirol. 1999, 5, 635–642. [Google Scholar] [CrossRef]
- Murray, P.D.; Krivacic, K.; Chernosky, A.; Wei, T.; Ransohoff, R.M.; Rodriguez, M. Biphasic and regionally-restricted chemokine expression in the central nervous system in the Theiler’s virus model of multiple sclerosis. J. Neurovirol. 2000, 6 (Suppl. 1), S44–S52. [Google Scholar] [PubMed]
- Theil, D.J.; Tsunoda, I.; Libbey, J.E.; Derfuss, T.J.; Fujinami, R.S. Alterations in cytokine but not chemokine mRNA expression during three distinct Theiler’s virus infections. J. Neuroimmunol. 2000, 104, 22–30. [Google Scholar] [CrossRef]
- Bennett, J.L.; Elhofy, A.; Charo, I.; Miller, S.D.; Dal Canto, M.C.; Karpus, W.J. CCR2 regulates development of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. Viral Immunol. 2007, 20, 19–33. [Google Scholar] [CrossRef]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Rawji, K.S.; Yong, V.W. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin. Dev. Immunol. 2013, 2013, 948976. [Google Scholar] [CrossRef]
- Giunti, D.; Parodi, B.; Cordano, C.; Uccelli, A.; Kerlero de Rosbo, N. Can we switch microglia’s phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology 2014, 141, 328–339. [Google Scholar] [CrossRef]
- Healy, L.M.; Michell-Robinson, M.A.; Antel, J.P. Regulation of human glia by multiple sclerosis disease modifying therapies. Semin. Immunopathol. 2015, 37, 639–649. [Google Scholar] [CrossRef]
- Kim, H.J.; Ifergan, I.; Antel, J.P.; Seguin, R.; Duddy, M.; Lapierre, Y.; Jalili, F.; Bar-Or, A. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J. Immunol. 2004, 172, 7144–7153. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Metz, L.M.; Li, D.; Traboulsee, A.; Myles, M.L.; Duquette, P.; Godin, J.; Constantin, M.; Yong, V.W. Glatiramer acetate in combination with minocycline in patients with relapsing--remitting multiple sclerosis: Results of a Canadian, multicenter, double-blind, placebo-controlled trial. Mult. Scler. 2009, 15, 1183–1194. [Google Scholar] [CrossRef]
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Trevick, S. The Epidemiology of Global Epilepsy. Neurol. Clin. 2016, 34, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Loscher, W.; Klein, P. The Pharmacology and Clinical Efficacy of Antiseizure Medications: From Bromide Salts to Cenobamate and Beyond. CNS Drugs 2021, 35, 935–963. [Google Scholar] [CrossRef]
- Perucca, E. From clinical trials of antiepileptic drugs to treatment. Epilepsia Open 2018, 3 (Suppl. S2), 220–230. [Google Scholar] [CrossRef]
- Perucca, E.; Brodie, M.J.; Kwan, P.; Tomson, T. 30 years of second-generation antiseizure medications: Impact and future perspectives. Lancet Neurol. 2020, 19, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef]
- Ahadiat, S.A.A.; Hosseinian, Z. SARS-CoV-2 and Seizure: An Insight Into the Pathophysiology. Anesthesiol. Pain Med. 2023, 13, e134129. [Google Scholar] [CrossRef] [PubMed]
- Annegers, J.F.; Hauser, W.A.; Beghi, E.; Nicolosi, A.; Kurland, L.T. The risk of unprovoked seizures after encephalitis and meningitis. Neurology 1988, 38, 1407–1410. [Google Scholar] [CrossRef]
- Getts, D.R.; Balcar, V.J.; Matsumoto, I.; Muller, M.; King, N.J. Viruses and the immune system: Their roles in seizure cascade development. J. Neurochem. 2008, 104, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Nem de Oliveira Souza, I.; Frost, P.S.; Franca, J.V.; Nascimento-Viana, J.B.; Neris, R.L.S.; Freitas, L.; Pinheiro, D.; Nogueira, C.O.; Neves, G.; Chimelli, L.; et al. Acute and chronic neurological consequences of early-life Zika virus infection in mice. Sci. Transl. Med. 2018, 10, eaar2749. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, D.; Oliveira, L.F.; Souza, I.N.O.; Brogin, J.A.F.; Bueno, D.D.; Miranda, I.A.; Da Poian, A.T.; Ferreira, S.T.; Figueiredo, C.P.; Clarke, J.R.; et al. Modulation in phase and frequency of neural oscillations during epileptiform activity induced by neonatal Zika virus infection in mice. Sci. Rep. 2020, 10, 6763. [Google Scholar] [CrossRef]
- Wu, H.M.; Huang, C.C.; Chen, S.H.; Liang, Y.C.; Tsai, J.J.; Hsieh, C.L.; Hsu, K.S. Herpes simplex virus type 1 inoculation enhances hippocampal excitability and seizure susceptibility in mice. Eur. J. Neurosci. 2003, 18, 3294–3304. [Google Scholar] [CrossRef]
- Metcalf, C.S.; Vanegas, F.; Underwood, T.; Johnson, K.; West, P.J.; Smith, M.D.; Wilcox, K.S. Screening of prototype antiseizure and anti-inflammatory compounds in the Theiler’s murine encephalomyelitis virus model of epilepsy. Epilepsia Open 2022, 7, 46–58. [Google Scholar] [CrossRef]
- Yamanaka, G.; Morichi, S.; Takamatsu, T.; Watanabe, Y.; Suzuki, S.; Ishida, Y.; Oana, S.; Yamazaki, T.; Takata, F.; Kawashima, H. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 4395. [Google Scholar] [CrossRef]
- Libbey, J.E.; Kennett, N.J.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Lack of correlation of central nervous system inflammation and neuropathology with the development of seizures following acute virus infection. J. Virol. 2011, 85, 8149–8157. [Google Scholar] [CrossRef]
- Stewart, K.A.; Wilcox, K.S.; Fujinami, R.S.; White, H.S. Development of postinfection epilepsy after Theiler’s virus infection of C57BL/6 mice. J. Neuropathol. Exp. Neurol. 2010, 69, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and brain excitability. Front. Neuroendocrinol. 2012, 33, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Pracucci, E.; Pillai, V.; Lamers, D.; Parra, R.; Landi, S. Neuroinflammation: A Signature or a Cause of Epilepsy? Int. J. Mol. Sci. 2021, 22, 6981. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef]
- Vezzani, A. Anti-inflammatory drugs in epilepsy: Does it impact epileptogenesis? Expert Opin. Drug Saf. 2015, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A. Brain Inflammation and Seizures: Evolving Concepts and New Findings in the Last 2 Decades. Epilepsy Curr. 2020, 20, 40S–43S. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Villasana-Salazar, B.; Vezzani, A. Neuroinflammation microenvironment sharpens seizure circuit. Neurobiol. Dis. 2023, 178, 106027. [Google Scholar] [CrossRef]
- Kirkman, N.J.; Libbey, J.E.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia 2010, 51, 454–464. [Google Scholar] [CrossRef]
- Broer, S.; Kaufer, C.; Haist, V.; Li, L.; Gerhauser, I.; Anjum, M.; Bankstahl, M.; Baumgartner, W.; Loscher, W. Brain inflammation, neurodegeneration and seizure development following picornavirus infection markedly differ among virus and mouse strains and substrains. Exp. Neurol. 2016, 279, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.L.; Heck, T.D.; Dahle, E.J.; Vanegas, F.; Pruess, T.H.; Wilcox, K.S.; White, H.S. Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia 2016, 57, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Cusick, M.F.; Libbey, J.E.; Doty, D.J.; DePaula-Silva, A.B.; Fujinami, R.S. The role of peripheral interleukin-6 in the development of acute seizures following virus encephalitis. J. Neurovirol. 2017, 23, 696–703. [Google Scholar] [CrossRef]
- de Vries, E.E.; van den Munckhof, B.; Braun, K.P.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. [Google Scholar] [CrossRef]
- Lorigados Pedre, L.; Morales Chacon, L.M.; Pavon Fuentes, N.; Robinson Agramonte, M.L.A.; Serrano Sanchez, T.; Cruz-Xenes, R.M.; Diaz Hung, M.L.; Estupinan Diaz, B.; Baez Martin, M.M.; Orozco-Suarez, S. Follow-Up of Peripheral IL-1beta and IL-6 and Relation with Apoptotic Death in Drug-Resistant Temporal Lobe Epilepsy Patients Submitted to Surgery. Behav. Sci. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Garcia-Oscos, F.; Salgado, H.; Hall, S.; Thomas, F.; Farmer, G.E.; Bermeo, J.; Galindo, L.C.; Ramirez, R.D.; D’Mello, S.; Rose-John, S.; et al. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol. Psychiatry 2012, 71, 574–582. [Google Scholar] [CrossRef]
- Mirabella, F.; Desiato, G.; Mancinelli, S.; Fossati, G.; Rasile, M.; Morini, R.; Markicevic, M.; Grimm, C.; Amegandjin, C.; Termanini, A.; et al. Prenatal interleukin 6 elevation increases glutamatergic synapse density and disrupts hippocampal connectivity in offspring. Immunity 2021, 54, 2611–2631 e2618. [Google Scholar] [CrossRef]
- Samland, H.; Huitron-Resendiz, S.; Masliah, E.; Criado, J.; Henriksen, S.J.; Campbell, I.L. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J. Neurosci. Res. 2003, 73, 176–187. [Google Scholar] [CrossRef]
- El-Haggar, S.M.; Hegazy, S.K.; Mustafa, W.; Khrieba, M.O. Possible immuno-modulatory effects of tocilizumab in patients with refractory status epilepticus. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Girardin, M.L.; Flamand, T.; Roignot, O.; Abi Warde, M.T.; Mutschler, V.; Voulleminot, P.; Guillot, M.; Dinkelacker, V.; De Saint-Martin, A. Treatment of new onset refractory status epilepticus/febrile infection-related epilepsy syndrome with tocilizumab in a child and a young adult. Epilepsia 2023, 64, e87–e92. [Google Scholar] [CrossRef]
- Kizilkilic, E.K.; Unkun, R.; Uygunoglu, U.; Delil, S.; Ozkara, C. Treatment of COVID-19-induced refractory status epilepticus by tocilizumab. Eur. J. Neurol. 2022, 29, 2861–2863. [Google Scholar] [CrossRef] [PubMed]
- Stredny, C.M.; Case, S.; Sansevere, A.J.; Son, M.; Henderson, L.; Gorman, M.P. Interleukin-6 Blockade With Tocilizumab in Anakinra-Refractory Febrile Infection-Related Epilepsy Syndrome (FIRES). Child. Neurol. Open 2020, 7, 2329048X20979253. [Google Scholar] [CrossRef] [PubMed]
- Monsour, M.; Croci, D.M.; Agazzi, S.; Borlongan, C.V. Contemplating IL-6, a double-edged sword cytokine: Which side to use for stroke pathology? CNS Neurosci. Ther. 2023, 29, 493–497. [Google Scholar] [CrossRef]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef]
- Aronica, E.; Gorter, J.A. Gene expression profile in temporal lobe epilepsy. Neuroscientist 2007, 13, 100–108. [Google Scholar] [CrossRef]
- Foresti, M.L.; Arisi, G.M.; Katki, K.; Montanez, A.; Sanchez, R.M.; Shapiro, L.A. Chemokine CCL2 and its receptor CCR2 are increased in the hippocampus following pilocarpine-induced status epilepticus. J. Neuroinflamm. 2009, 6, 40. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Mo, X.; Xi, Z.; Xiao, F.; Li, J.; Zhu, X.; Luan, G.; Wang, Y.; Li, Y.; et al. Expression of monocyte chemoattractant protein-1 in brain tissue of patients with intractable epilepsy. Clin. Neuropathol. 2008, 27, 55–63. [Google Scholar] [CrossRef]
- Kaufer, C.; Chhatbar, C.; Broer, S.; Waltl, I.; Ghita, L.; Gerhauser, I.; Kalinke, U.; Loscher, W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad. Sci. USA 2018, 115, E8929–E8938. [Google Scholar] [CrossRef]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef]
- Rossol, M.; Meusch, U.; Pierer, M.; Kaltenhauser, S.; Hantzschel, H.; Hauschildt, S.; Wagner, U. Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by contact with T cells. J. Immunol. 2007, 179, 4239–4248. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.M.S.; DePaula-Silva, A.B.; Doty, D.J.; Truong, A.; Libbey, J.E.; Fujinami, R.S. Microglial cell depletion is fatal with low level picornavirus infection of the central nervous system. J. Neurovirol. 2019, 25(3), 415–421. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, Y.; Wei, Y.; Bosco, D.B.; Xie, M.; Zhao, M.G.; Richardson, J.R.; Wu, L.J. Microglial depletion aggravates the severity of acute and chronic seizures in mice. Brain Behav. Immun. 2020, 89, 245–255. [Google Scholar] [CrossRef]
- Lei, F.; Cui, N.; Zhou, C.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R inhibition is not specific to innate immune cells but also affects T-helper cell differentiation independently of microglia depletion. Res. Sq. 2023, rs.3.rs-3308220. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
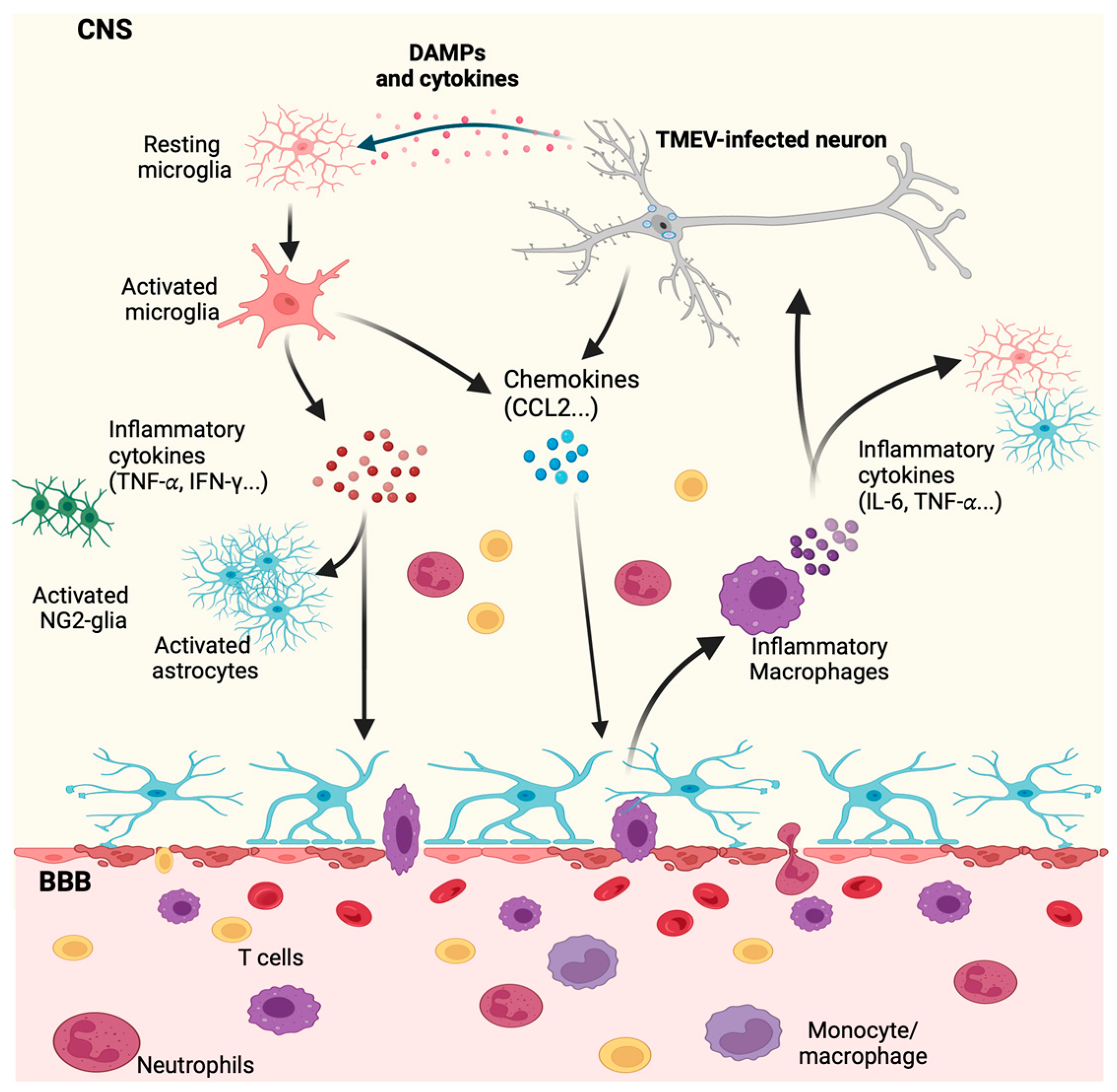
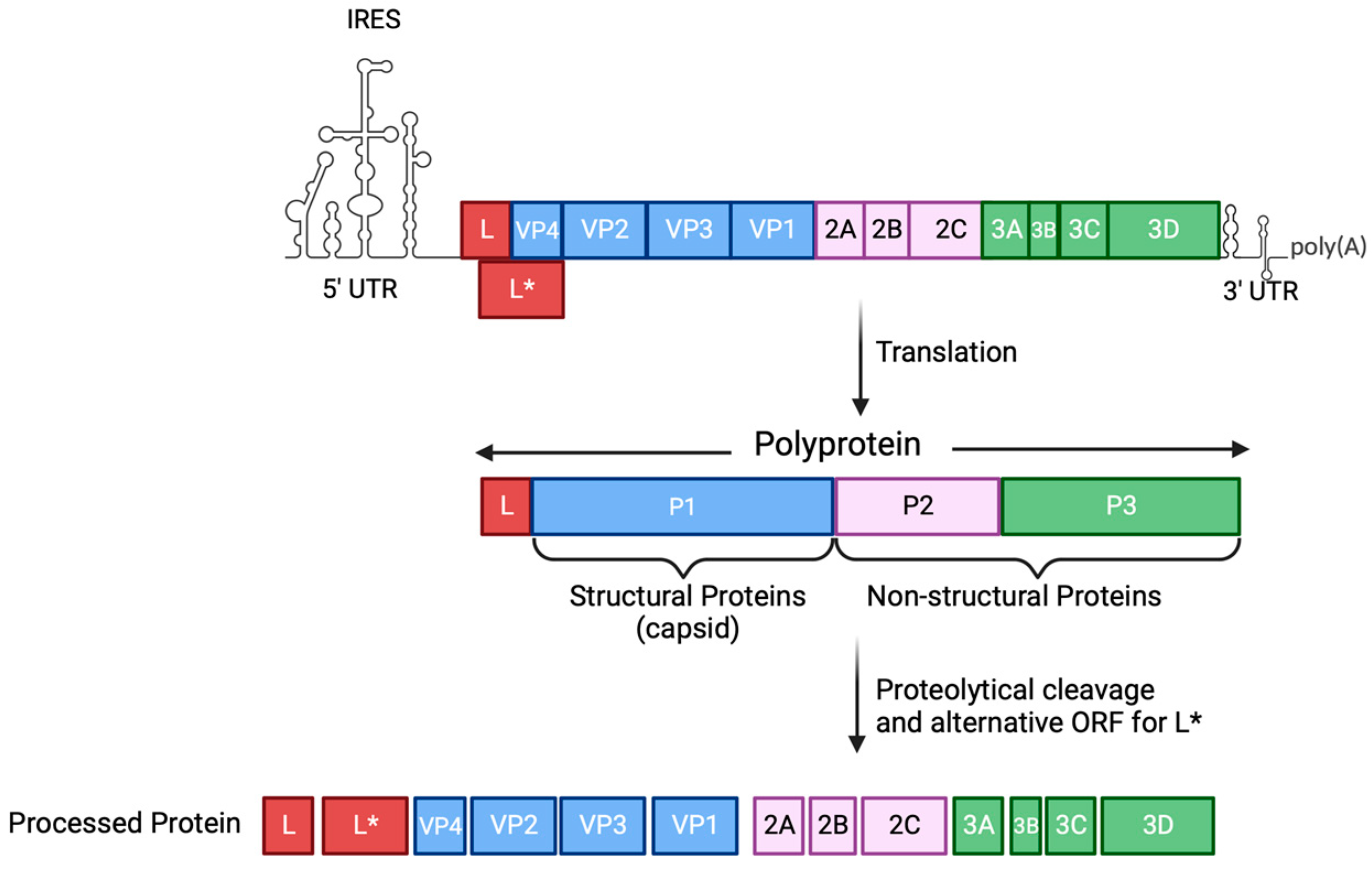
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DePaula-Silva, A.B. The Contribution of Microglia and Brain-Infiltrating Macrophages to the Pathogenesis of Neuroinflammatory and Neurodegenerative Diseases during TMEV Infection of the Central Nervous System. Viruses 2024, 16, 119. https://doi.org/10.3390/v16010119
DePaula-Silva AB. The Contribution of Microglia and Brain-Infiltrating Macrophages to the Pathogenesis of Neuroinflammatory and Neurodegenerative Diseases during TMEV Infection of the Central Nervous System. Viruses. 2024; 16(1):119. https://doi.org/10.3390/v16010119
Chicago/Turabian StyleDePaula-Silva, Ana Beatriz. 2024. "The Contribution of Microglia and Brain-Infiltrating Macrophages to the Pathogenesis of Neuroinflammatory and Neurodegenerative Diseases during TMEV Infection of the Central Nervous System" Viruses 16, no. 1: 119. https://doi.org/10.3390/v16010119
APA StyleDePaula-Silva, A. B. (2024). The Contribution of Microglia and Brain-Infiltrating Macrophages to the Pathogenesis of Neuroinflammatory and Neurodegenerative Diseases during TMEV Infection of the Central Nervous System. Viruses, 16(1), 119. https://doi.org/10.3390/v16010119