
Systematic Genomic Surveillance of SARS-CoV-2 at Xiamen International Airport and the Port of Xiamen Reveals the Importance of Incoming Travelers in Lineage Diversity

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample and Data Source
2.2. Next-Generation Sequencing
2.3. Phylogenetic and Evolution Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Bowe, B.; Xie, Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat. Med. 2022, 28, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Edara, V.-V.; Lai, L.; Sahoo, M.K.; Floyd, K.; Sibai, M.; Solis, D.; Flowers, M.W.; Hussaini, L.; Ciric, C.R.; Bechnack, S.; et al. Infection and vaccine-induced neutralizing antibody responses to the SARS-CoV-2 B.1.617.1 variant. bioRxiv 2021. bioRxiv:2021.05.09.443299. [Google Scholar]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Ito, J.; Zahradnik, J.; Fujita, S.; Kosugi, Y.; Schreiber, G.; Sato, K. Enhanced transmissibility, infectivity, and immune resistance of the SARS-CoV-2 omicron XBB.1.5 variant. Lancet Infect. Dis. 2023, 23, 280–281. [Google Scholar] [CrossRef]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023, 29, 344–347. [Google Scholar] [CrossRef]
- Butera, Y.; Mukantwari, E.; Artesi, M.; Umuringa, J.D.; O’Toole, Á.N.; Hill, V.; Rooke, S.; Hong, S.L.; Dellicour, S.; Majyambere, O.; et al. Genomic sequencing of SARS-CoV-2 in Rwanda reveals the importance of incoming travelers on lineage diversity. Nat. Commun. 2021, 12, 5705. [Google Scholar] [CrossRef]
- Olawoye, I.B.; Oluniyi, P.E.; Oguzie, J.U.; Uwanibe, J.N.; Kayode, T.A.; Olumade, T.J.; Ajogbasile, F.V.; Parker, E.; Eromon, P.E.; Abechi, P.; et al. Emergence and spread of two SARS-CoV-2 variants of interest in Nigeria. Nat. Commun. 2023, 14, 811. [Google Scholar] [CrossRef]
- Chen, S.; Huang, Z.; Guo, Y.; Guo, H.; Jian, L.; Xiao, J.; Yao, X.; Yu, H.; Cheng, T.; Zhang, Y.; et al. Evolving spike mutations in SARS-CoV-2 Omicron variants facilitate evasion from breakthrough infection-acquired antibodies. Cell Discov. 2023, 9, 86. [Google Scholar] [CrossRef]
- Moeti, M.; Gao, G.F.; Herrman, H. Global pandemic perspectives: Public health, mental health, and lessons for the future. Lancet 2022, 400, e3–e7. [Google Scholar] [CrossRef]
- Liu, J.; Liu, M.; Liang, W. The Dynamic COVID-Zero Strategy in China. China CDC Wkly 2022, 4, 74–75. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.-W.; Hu, B.; Chai, Y.; Yuen, T.T.-T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.-C.; Liu, H.; et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Chen, J.; Yang, K.; Zhang, M.; Shen, C.; Chen, J.; Wang, G.; Huang, S.; Zhang, M.; Xiong, H.; Chen, H.; et al. Rapid identification of imported influenza viruses at Xiamen International Airport via an active surveillance program. Clin. Microbiol. Infect. 2018, 24, 289–294. [Google Scholar] [CrossRef]
- Cowling, B.J.; Lau, L.L.H.; Wu, P.; Wong, H.W.C.; Fang, V.J.; Riley, S.; Nishiura, H. Entry screening to delay local transmission of 2009 pandemic influenza A (H1N1). BMC Infect. Dis. 2010, 10, 82. [Google Scholar] [CrossRef]
- Montalti, M.; Soldà, G.; Di Valerio, Z.; Salussolia, A.; Lenzi, J.; Forcellini, M.; Barvas, E.; Guttmann, S.; Messina, R.; Poluzzi, E.; et al. ROCCA observational study: Early results on safety of Sputnik V vaccine (Gam-COVID-Vac) in the Republic of San Marino using active surveillance. eClinicalMedicine 2021, 38, 101030. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 2022, 38, 1735–1737. [Google Scholar] [CrossRef]
- Tartof, S.Y.; Slezak, J.M.; Puzniak, L.; Hong, V.; Frankland, T.B.; Ackerson, B.K.; Xie, F.; Takhar, H.; Ogun, O.A.; Simmons, S.; et al. Effectiveness of BNT162b2 BA.4/5 bivalent mRNA vaccine against a range of COVID-19 outcomes in a large health system in the USA: A test-negative case–control study. Lancet Respir. Med. 2023, 11, 1089–1100. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, L.; Feng, Z.; Xu, H.; Li, F.; Shen, Y.; Zhang, D.; Liu, W.J.; Gao, G.F.; Wang, Q. Characterisation of SARS-CoV-2 variants in Beijing during 2022: An epidemiological and phylogenetic analysis. Lancet 2023, 401, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Ling, Y.; Jiang, M.; Tan, Y.; Wei, D.; Jiang, L.; Yu, S.; Jiang, F.; Wang, S.; Dai, Y.; et al. Primary assessment of the diversity of Omicron sublineages and the epidemiologic features of autumn/winter 2022 COVID-19 wave in Chinese mainland. Front. Med. 2023, 17, 758–767. [Google Scholar] [CrossRef]
- Ao, D.; He, X.; Hong, W.; Wei, X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm 2023, 4, e239. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Balakrishnan, K.N.; Yew, C.W.; Chong, E.T.; Daim, S.; Mohamad, N.E.; Rodrigues, K.; Lee, P.-C. Timeline of SARS-CoV-2 Transmission in Sabah, Malaysia: Tracking the Molecular Evolution. Pathogens 2023, 12, 1047. [Google Scholar] [CrossRef]
- Ramaiah, A.; Khubbar, M.; Akinyemi, K.; Bauer, A.; Carranza, F.; Weiner, J.; Bhattacharyya, S.; Payne, D.; Balakrishnan, N. Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA. Viruses 2023, 15, 1940. [Google Scholar] [CrossRef]
- Goh, A.X.C.; Chae, S.-R.; Chiew, C.J.; Tang, N.; Pang, D.; Lin, C.; Tan, K.B.; Lee, V.J.; Ho, Z.J.M. Characteristics of the omicron XBB subvariant wave in Singapore. Lancet 2023, 401, 1261–1262. [Google Scholar] [CrossRef]
- Velavan, T.P.; Ntoumi, F.; Kremsner, P.G.; Lee, S.S.; Meyer, C.G. Emergence and geographic dominance of Omicron subvariants XBB/XBB.1.5 and BF.7—The public health challenges. Int. J. Infect. Dis. 2023, 128, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Nie, K.; Zhao, H.; Zhao, X.; Ye, B.; Wang, J.; Chen, C.; Wang, H.; Di, J.; Li, J.; et al. Eleven COVID-19 Outbreaks with Local Transmissions Caused by the Imported SARS-CoV-2 Delta VOC—China, July-August, 2021. China CDC Wkly. 2021, 3, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Chen, Z.; Yu, A.; Li, X.; Feng, Y.; Zhao, X.; Xu, W.; Su, X. The First Two Imported Cases of SARS-CoV-2 Omicron Variant—Tianjin Municipality, China, December 13, 2021. China CDC Wkly 2022, 4, 76–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhao, X.; Chen, Z.; Nie, K.; Yin, Z.; Xia, Y.; Wang, J.; Niu, P.; Ruhan, A.; Li, L.; et al. Genomic Surveillance for SARS-CoV-2 Variants of Concern from Imported COVID-19 Cases—The Mainland of China, 2021. China CDC Wkly. 2022, 4, 680–684. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, M.; Lin, W.; Dong, W.; Xu, J. Evolutionary analysis of Omicron variant BF.7 and BA.5.2 pandemic in China. J. Biosaf. Biosecurity 2023, 5, 14–20. [Google Scholar] [CrossRef]
- Owen, D. Covid-19: Infections climb globally as EG.5 variant gains ground. BMJ 2023, 382, p1900. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Y.; Xu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Wang, P.; Wang, J.; Liu, J.; Yu, L.; et al. Fast evolution of SARS-CoV-2 BA.2.86 to JN.1 under heavy immune pressure. Lancet Infect. Dis. 2023. [Google Scholar] [CrossRef]
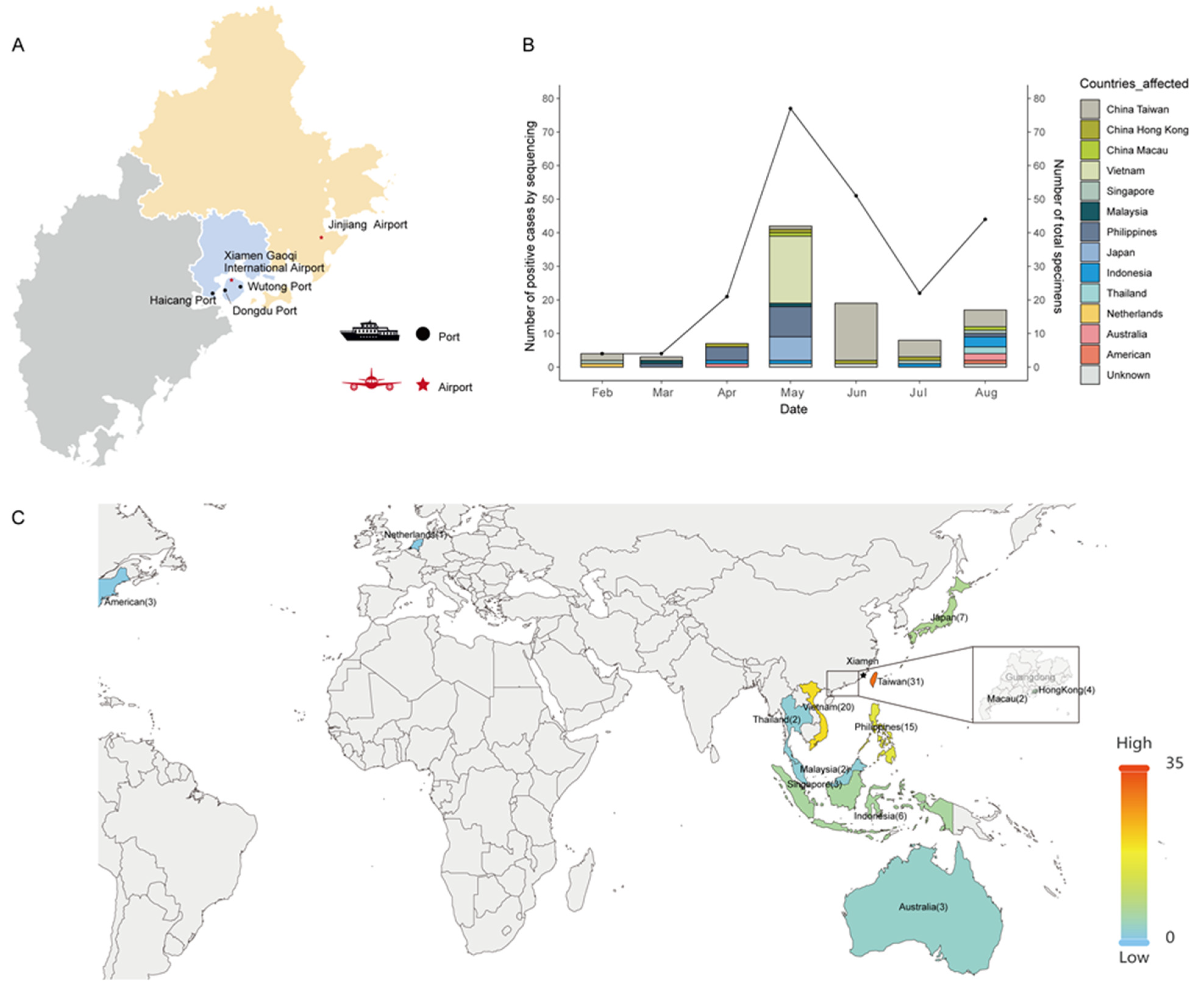
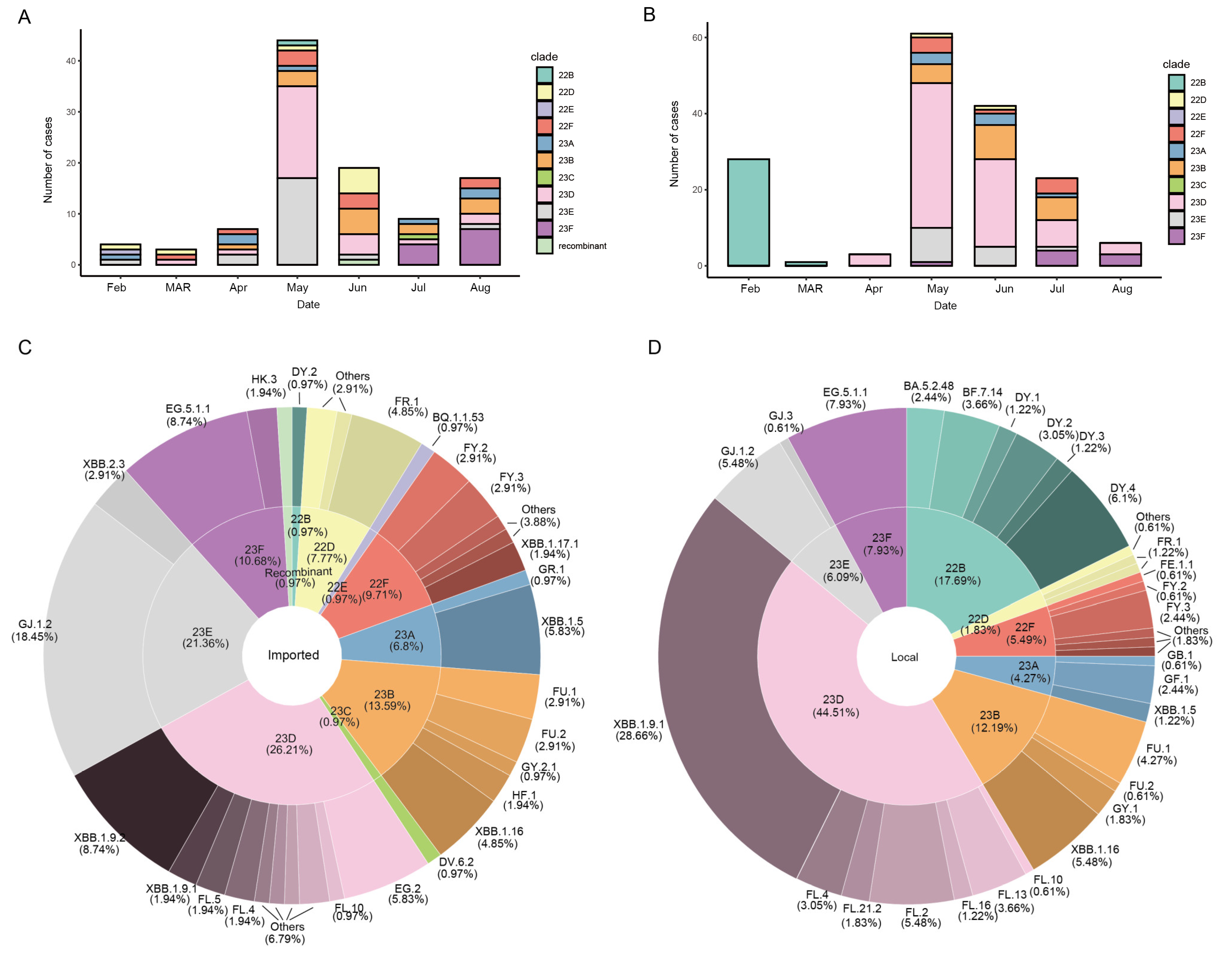


{kind=link}
{kind=link}
{kind=link}
| Country or Region Source | Clade (Numbers) | Date (Numbers) |
|---|---|---|
| America | 23D (1) | August |
| Australia | 23A (2) | April/August |
| 23F (1) | August | |
| China—Hong Kong | 23E (1) | April |
| 22B (1) | May | |
| 23B (1) | June | |
| 23F (1) | July | |
| China—Macau | 23A (1) | May |
| 23F (1) | August | |
| China—Taiwan | 22D (7) | February/March/June (5) |
| 22E (1) | February | |
| 22F (3) | June (3) | |
| 23A (2) | July/August | |
| 23B (6) | June (3)/July/August (2) | |
| 23C (1) | July | |
| 23D (6) | May/ June (4)/July | |
| 23E (1) | June | |
| 23F (3) | July/August (2) | |
| Recombinant | June | |
| Indonesia | 22F (1) | August |
| 23B (3) | April/July/August | |
| 23D (1) | August | |
| Japan | 23D (7) | May (7) |
| Malaysia | 23D (2) | March/May |
| 23B (1) | May | |
| The Netherlands | 23A (1) | February |
| The Philippines | 22F (3) | March/April /May |
| 23A (1) | April | |
| 23B (2) | May (2) | |
| 23D (3) | April /May (2) | |
| 23E (6) | April/May (4)/August | |
| Singapore | 23E (1) | February |
| 23F (2) | July/August | |
| Thailand | 22F (1) | August |
| 23F (1) | August | |
| Vietnam | 22F (2) | May (2) |
| 23D (5) | May (5) | |
| 23E (13) | May (13) | |
| Unknown | 22D (1) | May |
| 23B (1) | June | |
| 23F (1) | August |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, R.; Wu, R.; Wang, X.; Fu, R.; Xia, N.; Chen, Y.; Yang, K.; Chen, J. Systematic Genomic Surveillance of SARS-CoV-2 at Xiamen International Airport and the Port of Xiamen Reveals the Importance of Incoming Travelers in Lineage Diversity. Viruses 2024, 16, 132. https://doi.org/10.3390/v16010132
You R, Wu R, Wang X, Fu R, Xia N, Chen Y, Yang K, Chen J. Systematic Genomic Surveillance of SARS-CoV-2 at Xiamen International Airport and the Port of Xiamen Reveals the Importance of Incoming Travelers in Lineage Diversity. Viruses. 2024; 16(1):132. https://doi.org/10.3390/v16010132
Chicago/Turabian StyleYou, Ruiluan, Ruotong Wu, Xijing Wang, Rao Fu, Ningshao Xia, Yixin Chen, Kunyu Yang, and Junyu Chen. 2024. "Systematic Genomic Surveillance of SARS-CoV-2 at Xiamen International Airport and the Port of Xiamen Reveals the Importance of Incoming Travelers in Lineage Diversity" Viruses 16, no. 1: 132. https://doi.org/10.3390/v16010132