
The Inovirus Pf4 Triggers Antiviral Responses and Disrupts the Proliferation of Airway Basal Epithelial Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Basal Cell Culture
2.2. Single-Cell Sequencing
2.3. qRT-PCR
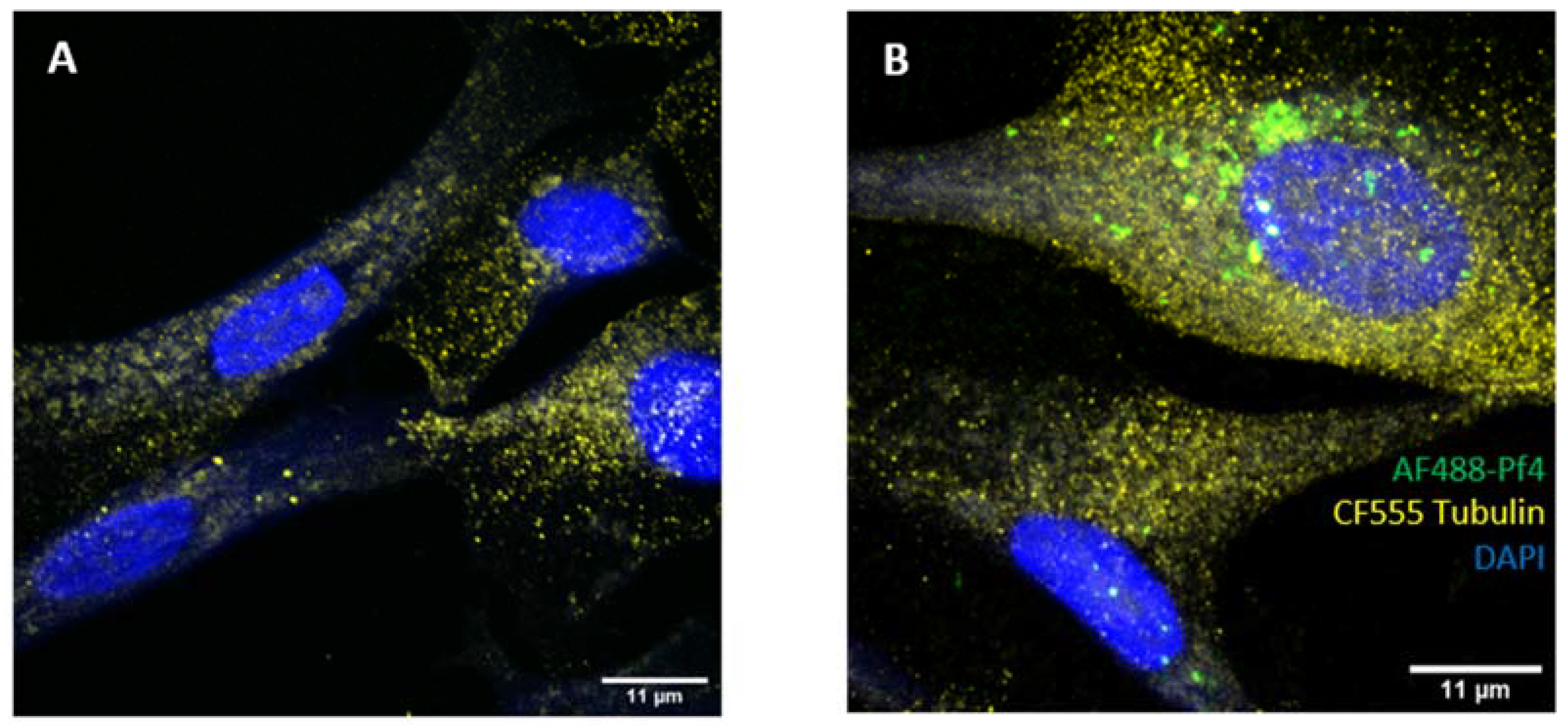
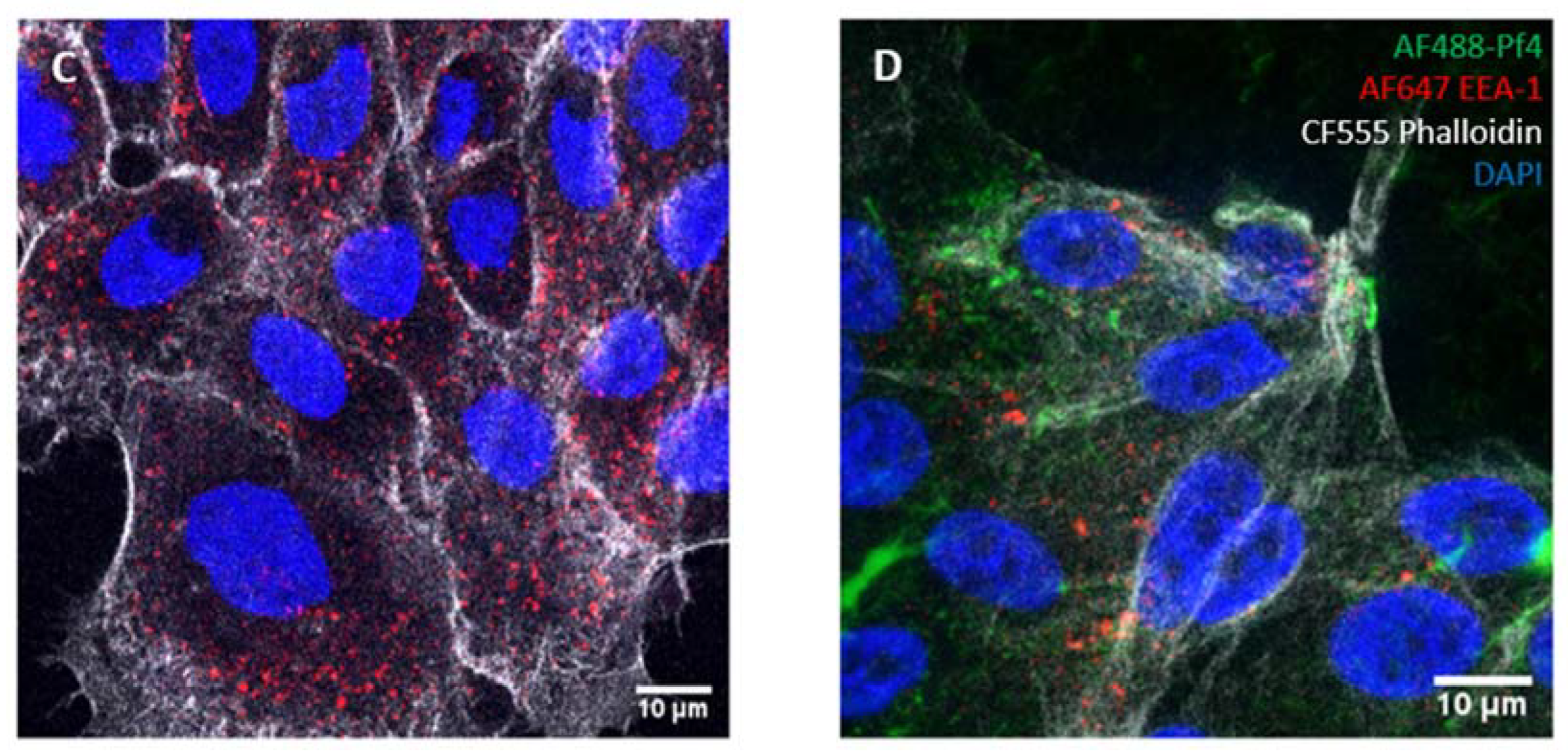
2.4. Microscopy
2.5. ELISA
2.6. Proliferation/Migration Assay
3. Results
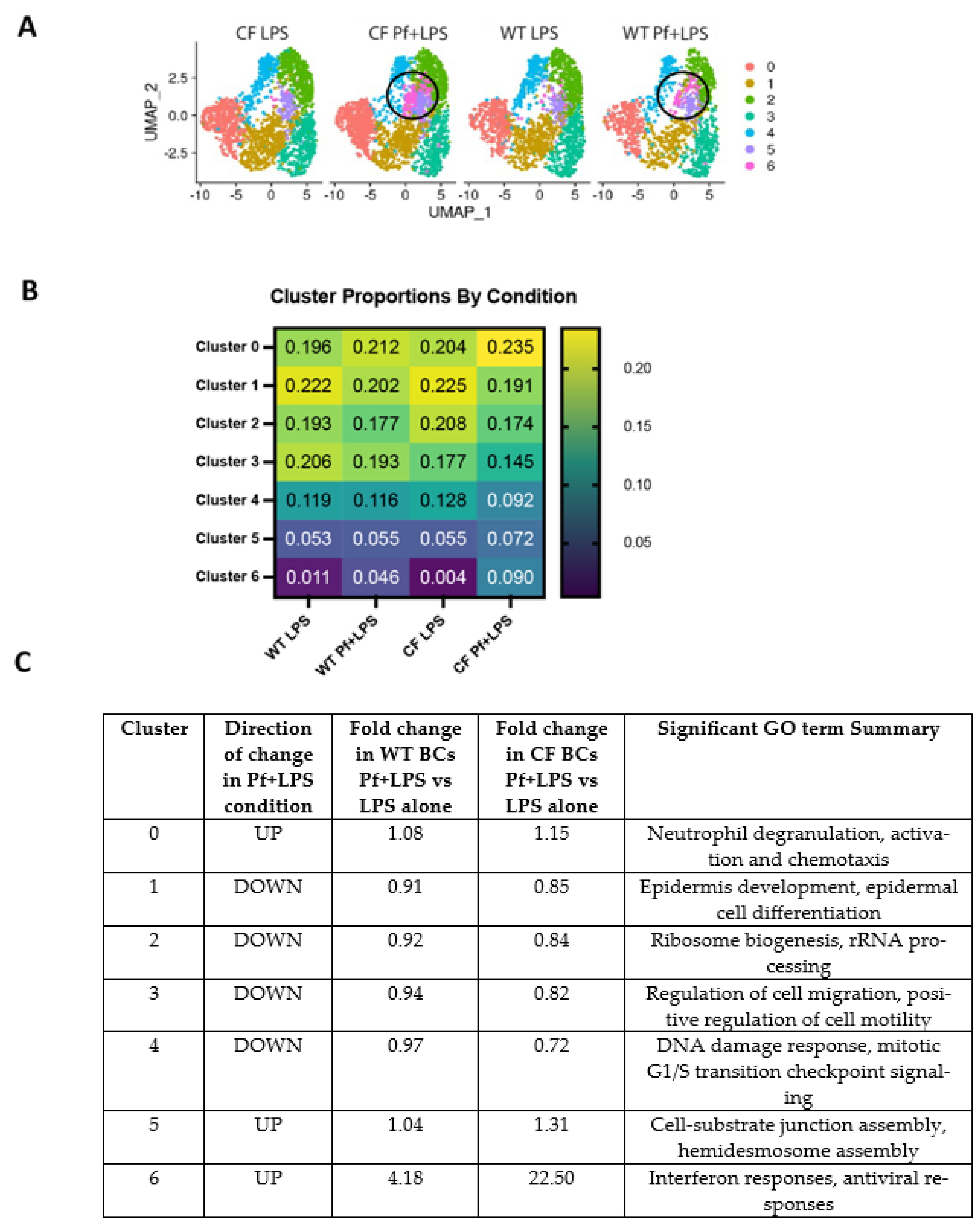
3.1. WT and CF BCs Cluster into Functionally Distinct Subsets
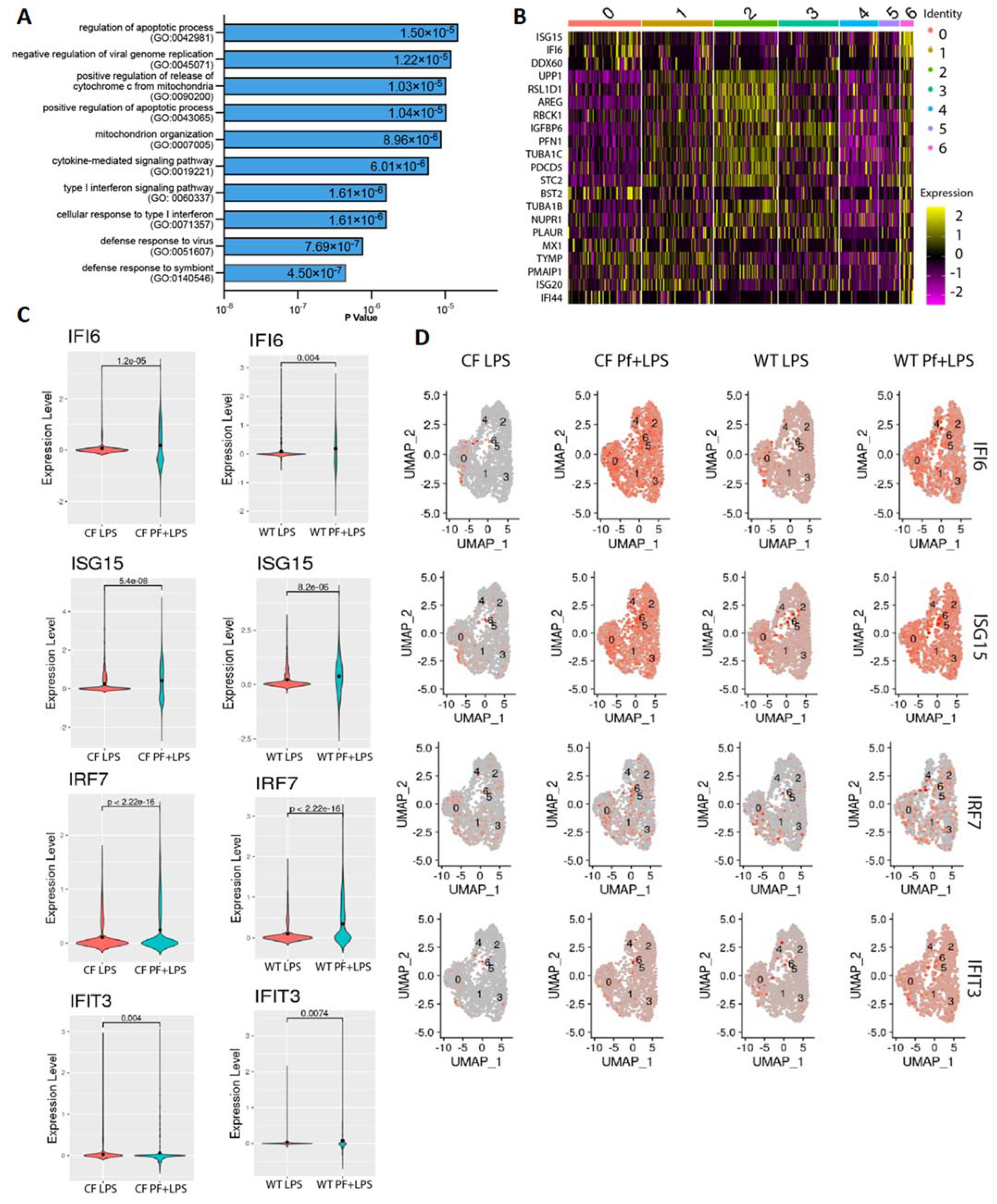
3.2. Pf4 Induces an Antiviral Response in a Subset of BCs
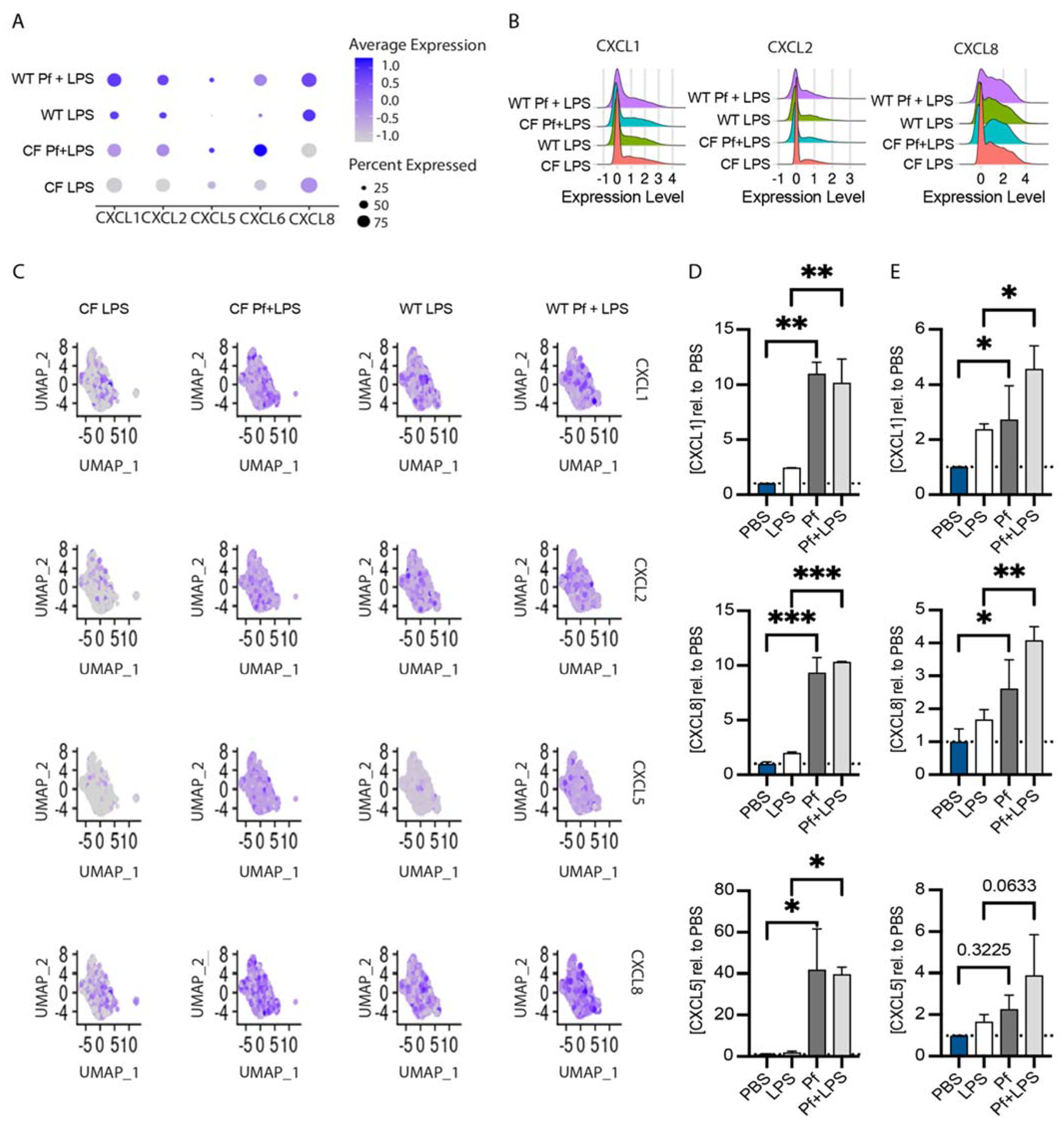
3.3. Pf4 Phage Increases Neutrophil Chemoattractant Production in BCs
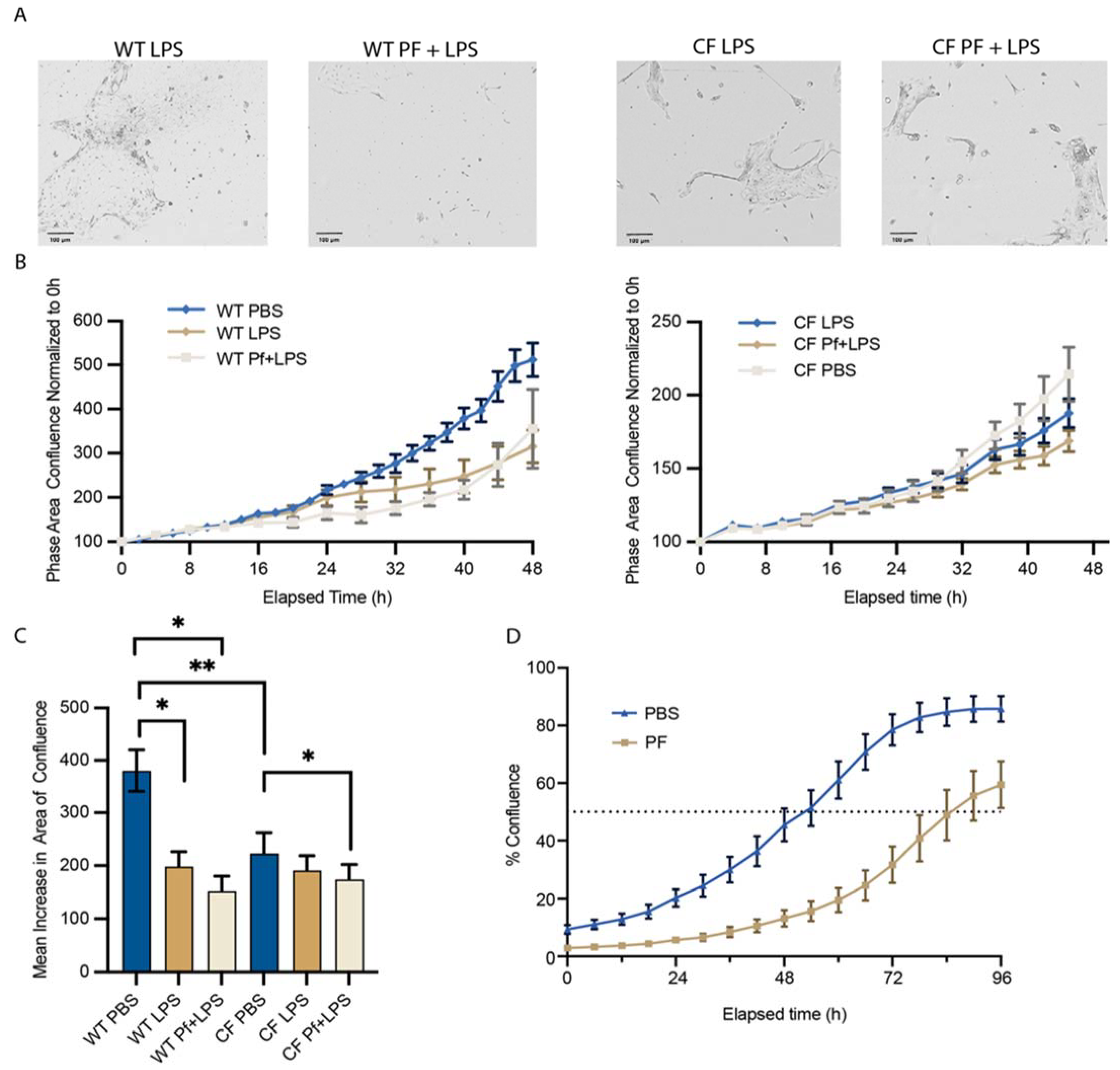
3.4. Pf4 Impairs the Ability of BCs to Reach Confluence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fauquet, C.M. (Ed.) The Single Stranded DNA Viruses, in Virus Taxonomy; Academic Press: San Diego, CA, USA, 2005; pp. 277–369. [Google Scholar]
- Hay, I.D.; Lithgow, T. Filamentous phages: Masters of a microbial sharing economy. EMBO Rep. 2019, 20, e47427. [Google Scholar] [CrossRef] [PubMed]
- Mai-Prochnow, A.; Hui, J.G.K.; Kjelleberg, S.; Rakonjac, J.; McDougald, D.; Rice, S.A. Big things in small packages: The genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol. Rev. 2015, 39, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, P.; Voet, M.; Lavigne, R. Prevalence of Pf1-like (pro)phage genetic elements among Pseudomonas aeruginosa isolates. Virology 2015, 483, 64–71. [Google Scholar] [CrossRef]
- Secor, P.R.; Burgener, E.B.; Kinnersley, M.; Jennings, L.K.; Roman-Cruz, V.; Popescu, M.; Van Belleghem, J.D.; Haddock, N.; Copeland, C.; Michaels, L.A.; et al. Pf Bacteriophage and Their Impact on Pseudomonas Virulence, Mammalian Immunity, and Chronic Infections. Front. Immunol. 2020, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry. 2021 Annual Data Report; US Cystic Fibrosis Foundation, Ed.; Bethesda: Rockville, MD, USA, 2022. [Google Scholar]
- Nixon, G.M.; Armstrong, D.S.; Carzino, R.; Carlin, J.B.; Olinsky, A.; Robertson, C.F.; Grimwood, K. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J. Pediatr. 2001, 138, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.; Gibson, R.L.; MN, S.M.; Emerson, J.; Burns, J.L.; Castile, R.; Hiatt, P.; McCoy, K.; Wilson, C.B.; Inglis, A.; et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr. Pulmonol. 2001, 32, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Wagener, J.S.; VanDevanter, D.R. Characterizing aggressiveness and predicting future progression of CF lung disease. J. Cyst. Fibros. 2009, 8, S15–S19. [Google Scholar] [CrossRef]
- Percival, S.L.; Suleman, L.; Vuotto, C.; Donelli, G. Healthcare-associated infections, medical devices and biofilms: Risk, tolerance and control. J. Med Microbiol. 2015, 64, 323–334. [Google Scholar] [CrossRef]
- Hirsch, E.B.; Tam, V.H. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev. Pharmacoecon. Outcomes Res. 2010, 10, 441–451. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Burgener, E.B.; Sweere, J.M.; Bach, M.S.; Secor, P.R.; Haddock, N.; Jennings, L.K.; Marvig, R.L.; Johansen, H.K.; Rossi, E.; Cao, X.; et al. Filamentous bacteriophages are associated with chronic Pseudomonas lung infections and antibiotic resistance in cystic fibrosis. Sci. Transl. Med. 2019, 11, 488. [Google Scholar] [CrossRef] [PubMed]
- Burgener, E.B.; Secor, P.R.; Tracy, M.C.; Sweere, J.M.; Bik, E.M.; Milla, C.E.; Bollyky, P.L. Methods for Extraction and Detection of Pf Bacteriophage DNA from the Sputum of Patients with Cystic Fibrosis. PHAGE 2020, 1, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Fiedoruk, K.; Zakrzewska, M.; Daniluk, T.; Piktel, E.; Chmielewska, S.; Bucki, R. Two Lineages of Pseudomonas aeruginosa Filamentous Phages: Structural Uniformity over Integration Preferences. Genome Biol. Evol. 2020, 12, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Carnevali, P.B.M.; Cheng, J.-F.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Marvin, D.; Symmons, M.; Straus, S. Structure and assembly of filamentous bacteriophages. Prog. Biophys. Mol. Biol. 2014, 114, 80–122. [Google Scholar] [CrossRef]
- Secor, P.R.; Sweere, J.M.; Michaels, L.A.; Malkovskiy, A.V.; Lazzareschi, D.; Katznelson, E.; Rajadas, J.; Birnbaum, M.E.; Arrigoni, A.; Braun, K.R.; et al. Filamentous Bacteriophage Promote Biofilm Assembly and Function. Cell Host Microbe 2015, 18, 549–559. [Google Scholar] [CrossRef]
- Tarafder, A.K.; von Kügelgen, A.; Mellul, A.J.; Schulze, U.; Aarts, D.G.A.L.; Bharat, T.A.M. Phage liquid crystalline droplets form occlusive sheaths that encapsulate and protect infectious rod-shaped bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 4724–4731. [Google Scholar] [CrossRef]
- Secor, P.; Jennings, L.; Michaels, L.; Sweere, J.; Singh, P.; Parks, W.; Bollyky, P. Biofilm assembly becomes crystal clear—filamentous bacteriophage organize the Pseudomonas aeruginosa biofilm matrix into a liquid crystal. Microb. Cell 2016, 3, 49–52. [Google Scholar] [CrossRef]
- Böhning, J.; Graham, M.; Letham, S.C.; Davis, L.K.; Schulze, U.; Stansfeld, P.J.; Corey, R.A.; Pearce, P.; Tarafder, A.K.; Bharat, T.A.M. Biophysical basis of filamentous phage tactoid-mediated antibiotic tolerance in P. aeruginosa. Nat. Commun. 2023, 14, 1–15. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Tang, K.; Wang, P.; Zeng, Z.; Guo, Y.; Wang, X. Excisionase in Pf filamentous prophage controls lysis-lysogeny decision-making in Pseudomonas aeruginosa. Mol. Microbiol. 2019, 111, 495–513. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.H.; Michie, K.A.; Goh, Y.F.; Noorian, P.; Kjelleberg, S.; Duggin, I.G.; McDougald, D.; Rice, S.A. The Repressor C Protein, Pf4r, Controls Superinfection of Pseudomonas aeruginosa PAO1 by the Pf4 Filamentous Phage and Regulates Host Gene Expression. Viruses 2021, 13, 1614. [Google Scholar] [CrossRef] [PubMed]
- Secor, P.R.; Michaels, L.A.; Smigiel, K.S.; Rohani, M.G.; Jennings, L.K.; Hisert, K.B.; Arrigoni, A.; Braun, K.R.; Birkland, T.P.; Lai, Y.; et al. Filamentous Bacteriophage Produced by Pseudomonas aeruginosa Alters the Inflammatory Response and Promotes Noninvasive Infection In Vivo. Infect. Immun. 2017, 85, 100–128. [Google Scholar] [CrossRef] [PubMed]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef] [PubMed]
- Gavric, D.; Knezevic, P. Filamentous Pseudomonas Phage Pf4 in the Context of Therapy-Inducibility, Infectivity, Lysogenic Conversion, and Potential Application. Viruses 2022, 14, 1261. [Google Scholar] [CrossRef] [PubMed]
- Tortuel, D.; Tahrioui, A.; David, A.; Cambronel, M.; Nilly, F.; Clamens, T.; Maillot, O.; Barreau, M.; Feuilloley, M.G.J.; Lesouhaitier, O.; et al. Pf4 Phage Variant Infection Reduces Virulence-Associated Traits in Pseudomonas aeruginosa. Microbiol. Spectr. 2022, 10, e0154822. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef]
- Wansleeben, C.; Barkauskas, C.E.; Rock, J.R.; Hogan, B.L.M. Stem cells of the adult lung: Their development and role in homeostasis, regeneration, and disease. Wiley Interdiscip. Rev. Dev. Biol. 2012, 2, 131–148. [Google Scholar] [CrossRef]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef]
- Parker, D.; Prince, A. Innate Immunity in the Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef]
- Carraro, G.; Langerman, J.; Sabri, S.; Lorenzana, Z.; Purkayastha, A.; Zhang, G.; Konda, B.; Aros, C.J.; Calvert, B.A.; Szymaniak, A.; et al. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med. 2021, 27, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Sanmukh, S.G.; dos Santos, N.J.; Barquilha, C.N.; Cucielo, M.S.; de Carvalho, M.; dos Reis, P.P.; Delella, F.K.; Carvalho, H.F.; Felisbino, S.L. Bacteriophages M13 and T4 Increase the Expression of Anchorage-Dependent Survival Pathway Genes and Down Regulate Androgen Receptor Expression in LNCaP Prostate Cell Line. Viruses 2021, 13, 1754. [Google Scholar] [CrossRef] [PubMed]
- Sanmukh, S.G.; Felisbino, S.L. Development of pipette tip gap closure migration assay (s-ARU method) for studying semi-adherent cell lines. Cytotechnology 2018, 70, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Møller-Olsen, C.; Ho, S.F.S.; Shukla, R.D.; Feher, T.; Sagona, A.P. Engineered K1F bacteriophages kill intracellular Escherichia coli K1 in human epithelial cells. Sci. Rep. 2018, 8, 17559. [Google Scholar] [CrossRef] [PubMed]
- Vladar, E.K.; Kunimoto, K.; Rojas-Hernandez, L.S.; Spano, J.M.; Sellers, Z.M.; Joo, N.S.; Cooney , R.A.; Axelrod, J.D.; Milla, C.E. Notch signaling inactivation by small molecule gamma-secretase inhibitors restores the multiciliated cell population in the airway epithelium. Am. J. Physiol. Lung. Cell Mol. Physiol. 2023, 324, L771–L782. [Google Scholar] [CrossRef]
- Vladar, E.K.; Nayak, J.V.; Milla, C.E.; Axelrod, J.D. Airway epithelial homeostasis and planar cell polarity signaling depend on multiciliated cell differentiation. J. Clin. Investig. 2016, 1, e88027. [Google Scholar] [CrossRef]
- Cady, K.C.; Bondy-Denomy, J.; Heussler, G.E.; Davidson, A.R.; O’Toole, G.A. The CRISPR/Cas Adaptive Immune System of Pseudomonas aeruginosa Mediates Resistance to Naturally Occurring and Engineered Phages. J. Bacteriol. 2012, 194, 5728–5738. [Google Scholar] [CrossRef]
- Lee, C.-H.; Frasch, C.E. Quantification of Bacterial Polysaccharides by the Purpald Assay: Measurement of Periodate-Generated Formaldehyde from Glycol in the Repeating Unit. Anal. Biochem. 2001, 296, 73–82. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.V.; Yemaneab, B.T.; Jass, J.; Scherbak, N. Reference Gene Selection for qPCR Is Dependent on Cell Type Rather than Treatment in Colonic and Vaginal Human Epithelial Cell Lines. PLoS ONE 2014, 9, e115592. [Google Scholar] [CrossRef]
- Varenyiova, Z.; Rojas-Hernandez, L.S.; Spano, J.; Capek, V.; Rosenberg-Hasson, Y.; Holmes, T.; Milla, C. Azithromycin promotes proliferation, and inhibits inflammation in nasal epithelial cells in primary ciliary dyskinesia. Sci. Rep. 2023, 13, 14453. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Amiri, M.; Sonenberg, N. Translational Control in Stem Cells. Front. Genet. 2019, 9, 709. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J. A bacteriophages journey through the human body. Immunol. Rev. 2017, 279, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Bichet, M.C.; Adderley, J.; Avellaneda-Franco, L.; Magnin-Bougma, I.; Torriero-Smith, N.; Gearing, L.J.; Deffrasnes, C.; David, C.; Pepin, G.; Gantier, M.P.; et al. Mammalian cells internalize bacteriophages and use them as a resource to enhance cellular growth and survival. PLoS Biol. 2023, 21, e3002341. [Google Scholar] [CrossRef] [PubMed]
- Jakiela, B.; Brockman-Schneider, R.; Amineva, S.; Lee, W.-M.; Gern, J.E. Basal Cells of Differentiated Bronchial Epithelium Are More Susceptible to Rhinovirus Infection. Am. J. Respir. Cell Mol. Biol. 2008, 38, 517–523. [Google Scholar] [CrossRef]
- Persson, B.D.; Jaffe, A.B.; Fearns, R.; Danahay, H. Respiratory Syncytial Virus Can Infect Basal Cells and Alter Human Airway Epithelial Differentiation. PLoS ONE 2014, 9, e102368. [Google Scholar] [CrossRef]
- Hewson, C.A.; Jardine, A.; Edwards, M.R.; Laza-Stanca, V.; Johnston, S.L. Toll-Like Receptor 3 Is Induced by and Mediates Antiviral Activity against Rhinovirus Infection of Human Bronchial Epithelial Cells. J. Virol. 2005, 79, 12273–12279. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Kirsebom, F.C.M. Neutrophils in respiratory viral infections. Mucosal Immunol. 2021, 14, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.M.; Douglass, M.F.; Chatterjee, D.; Akira, S.; Baaten, B.J.G. Matrix Metalloprotease 9 Mediates Neutrophil Migration into the Airways in Response to Influenza Virus-Induced Toll-Like Receptor Signaling. PLoS Pathog. 2012, 8, e1002641. [Google Scholar] [CrossRef] [PubMed]
- Kurzepa-Skaradzinska, A.; Skaradzinski, G.; Weber-Dabrowska, B.; Zaczek, M.; Maj, T.; Slawek, A.; Switalska, M.; Maciejewska, M.; Wietrzyk, J.; Rymowicz, W.; et al. Influence of bacteriophage preparations on migration of HL-60 leukemia cells in vitro. Anticancer Res. 2013, 33, 1569–1574. [Google Scholar] [PubMed]
- Dąbrowska, K.; Skaradziński, G.; Jończyk, P.; Kurzępa, A.; Wietrzyk, J.; Owczarek, B.; Żaczek, M.; Świtała-Jeleń, K.; Boratyński, J.; Poźniak, G.; et al. The effect of bacteriophages T4 and HAP1 on in vitro melanoma migration. BMC Microbiol. 2009, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Ali, Z.S.; Sweezey, N.; Grasemann, H.; Palaniyar, N. Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes 2019, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Yonker, L.M.; Cigana, C.; Hurley, B.P.; Bragonzi, A. Host-pathogen interplay in the respiratory environment of cystic fibrosis. J. Cyst. Fibros. 2015, 14, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Gaggar, A.; Bruscia, E.; Hector, A.; Marcos, V.; Jung, A.; Greene, C.; McElvaney, G.; Mall, M.; Döring, G. Innate immunity in cystic fibrosis lung disease. J. Cyst. Fibros. 2012, 11, 363–382. [Google Scholar] [CrossRef]
- Petersen, N.T.; Høiby, N.; Mordhorst, C.H.; Lind, K.; Flensborg, E.W.; Bruun, B. Respiratory infections in cystic fibrosis patients caused by virus, chlamydia and mycoplasma--possible synergism with Pseudomonas aeruginosa. Acta Paediatr. Scand. 1981, 70, 623–628. [Google Scholar] [CrossRef]
- Chattoraj, S.S.; Ganesan, S.; Jones, A.M.; Helm, J.M.; Comstock, A.T.; Bright-Thomas, R.; LiPuma, J.J.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus infection liberates planktonic bacteria from biofilm and increases chemokine responses in cystic fibrosis airway epithelial cells. Thorax 2011, 66, 333–339. [Google Scholar] [CrossRef]
- Hendricks, M.R.; Lashua, L.P.; Fischer, D.K.; Flitter, B.A.; Eichinger, K.M.; Durbin, J.E.; Sarkar, S.N.; Coyne, C.B.; Empey, K.M.; Bomberger, J.M. Respiratory syncytial virus infection enhances Pseudomonas aeruginosa biofilm growth through dysregulation of nutritional immunity. Proc. Natl. Acad. Sci. USA 2016, 113, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Manohar, P.; Loh, B.; Nachimuthu, R.; Hua, X.; Welburn, S.C.; Leptihn, S. Secondary Bacterial Infections in Patients with Viral Pneumonia. Front. Med. 2020, 7, 420. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.K.; Rau, M.H.; Johansen, H.K.; Ciofu, O.; Jelsbak, L.; Yang, L.; Folkesson, A.; Jarmer, H.; Aanæs, K.; von Buchwald, C.; et al. Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J. 2011, 6, 31–45. [Google Scholar] [CrossRef] [PubMed]
- McDougall, C.M.; Blaylock, M.G.; Douglas, J.G.; Brooker, R.J.; Helms, P.J.; Walsh, G.M. Nasal Epithelial Cells as Surrogates for Bronchial Epithelial Cells in Airway Inflammation Studies. Am. J. Respir. Cell Mol. Biol. 2008, 39, 560–568. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popescu, M.C.; Haddock, N.L.; Burgener, E.B.; Rojas-Hernandez, L.S.; Kaber, G.; Hargil, A.; Bollyky, P.L.; Milla, C.E. The Inovirus Pf4 Triggers Antiviral Responses and Disrupts the Proliferation of Airway Basal Epithelial Cells. Viruses 2024, 16, 165. https://doi.org/10.3390/v16010165
Popescu MC, Haddock NL, Burgener EB, Rojas-Hernandez LS, Kaber G, Hargil A, Bollyky PL, Milla CE. The Inovirus Pf4 Triggers Antiviral Responses and Disrupts the Proliferation of Airway Basal Epithelial Cells. Viruses. 2024; 16(1):165. https://doi.org/10.3390/v16010165
Chicago/Turabian StylePopescu, Medeea C., Naomi L. Haddock, Elizabeth B. Burgener, Laura S. Rojas-Hernandez, Gernot Kaber, Aviv Hargil, Paul L. Bollyky, and Carlos E. Milla. 2024. "The Inovirus Pf4 Triggers Antiviral Responses and Disrupts the Proliferation of Airway Basal Epithelial Cells" Viruses 16, no. 1: 165. https://doi.org/10.3390/v16010165
APA StylePopescu, M. C., Haddock, N. L., Burgener, E. B., Rojas-Hernandez, L. S., Kaber, G., Hargil, A., Bollyky, P. L., & Milla, C. E. (2024). The Inovirus Pf4 Triggers Antiviral Responses and Disrupts the Proliferation of Airway Basal Epithelial Cells. Viruses, 16(1), 165. https://doi.org/10.3390/v16010165