
Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage
 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Ethics
2.3. Molecular Investigation
2.4. Whole-Genome Sequencing
2.5. Genome Assembling and Variant Analyses
2.6. Evolutionary Analyses
2.7. Statistics
3. Results
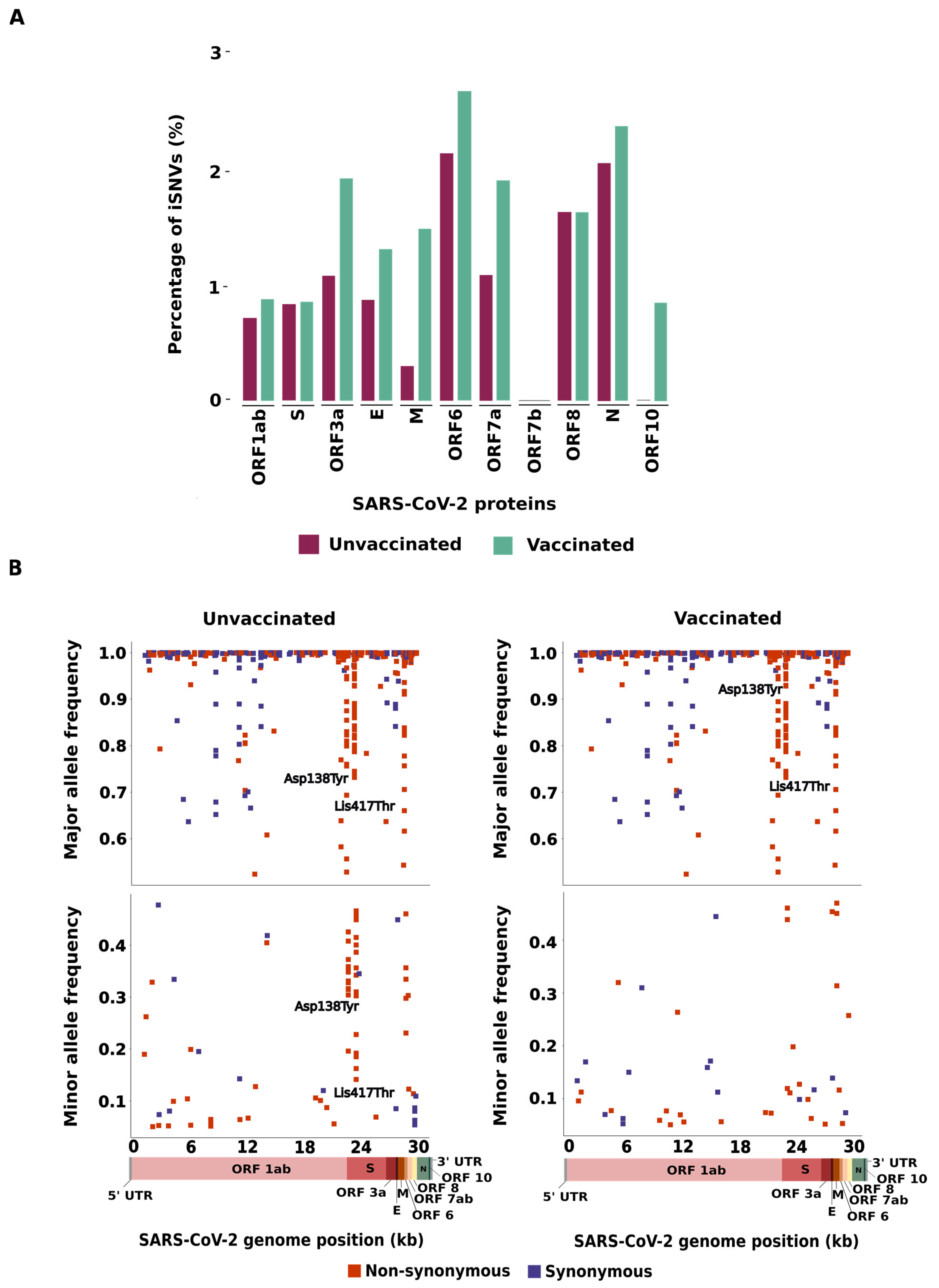
3.1. Distribution of iSNVs across SARS-CoV-2 Genomes
3.2. Characterization of the iSNVs Detected in the SARS-CoV-2 Genomes
3.3. Allele Composition of the SARS-CoV-2 Genomes
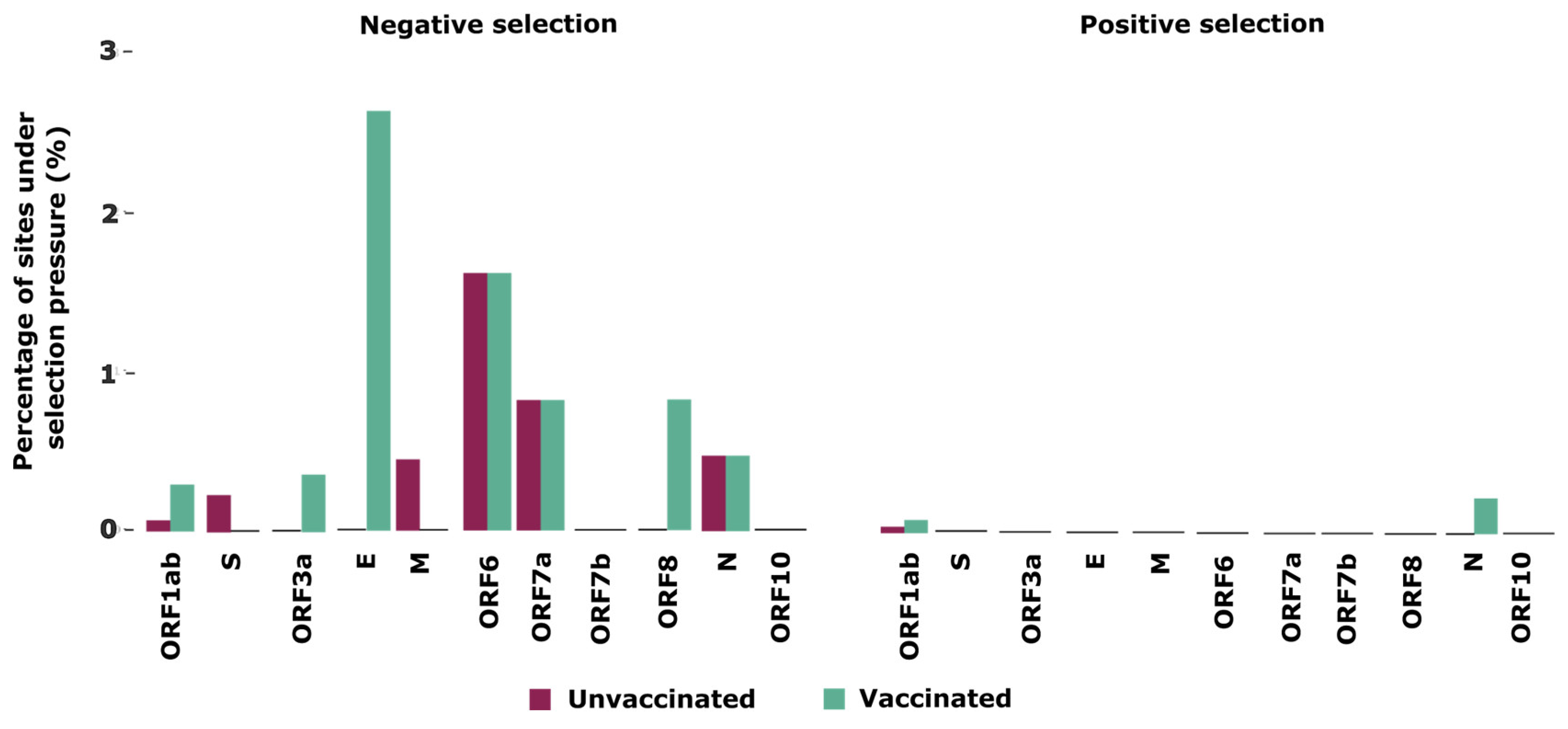
3.4. Selection Pressures Detected in the SARS-CoV-2 Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; da Candido, D.S.; Mishra, S.; E Crispim, M.A.; S Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Ciccozzi, M.; Zella, D.; Bianchi, M.; Benedetti, F.; Benvenuto, D.; Broccolo, F.; Cauda, R.; Caruso, A.; Angeletti, S.; et al. SARS-CoV-2 B.1.617 Indian Variants: Are Electrostatic Potential Changes Responsible for a Higher Transmission Rate? J. Med. Virol. 2021, 93, 6551–6556. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural Biology of SARS-CoV-2 and Implications for Therapeutic Development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Kashte, S.; Gulbake, A.; El-Amin, S.F.; Gupta, A. COVID-19 Vaccines: Rapid Development, Implications, Challenges and Future Prospects. Hum. Cell 2021, 34, 711–733. [Google Scholar] [CrossRef]
- Li, R.; Liu, J.; Zhang, H. The Challenge of Emerging SARS-CoV-2 Mutants to Vaccine Development. J. Genet. Genom. 2021, 48, 102–106. [Google Scholar] [CrossRef]
- Banho, C.A.; Sacchetto, L.; Campos, G.R.F.; Bittar, C.; Possebon, F.S.; Ullmann, L.S.; Marques, B.d.C.; da Silva, G.C.D.; Moraes, M.M.; Parra, M.C.P.; et al. Impact of SARS-CoV-2 Gamma Lineage Introduction and COVID-19 Vaccination on the Epidemiological Landscape of a Brazilian City. Commun. Med. 2022, 2, 41. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; et al. Vaccine Breakthrough Infections with SARS-CoV-2 Variants. N. Engl. J. Med. 2021, 384, 2212–2218. [Google Scholar] [CrossRef]
- Estofolete, C.F.; Banho, C.A.; Campos, G.R.F.; Marques, B.C.; Sacchetto, L.; Ullmann, L.S.; Possebon, F.S.; Machado, L.F.; Syrio, J.D.; Araújo Junior, J.P.; et al. Case Study of Two Post Vaccination SARS-CoV-2 Infections with P1 Variants in Coronavac Vaccinees in Brazil. Viruses 2021, 13, 1237. [Google Scholar] [CrossRef]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 Exhibits Intra-Host Genomic Plasticity and Low-Frequency Polymorphic Quasispecies. J. Clin. Virol. 2020, 131, 104585. [Google Scholar] [CrossRef] [PubMed]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-Host Diversity and Transmission. Science 2021, 372, eabg0821. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.C. Intra-Host Diversity of Sars-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; Da Silva Francisco, R.; De Almeida, L.G.P.; Brustolini, O.J.; Cardoso, C.C.; Gerber, A.L.; Guimarães, A.P.D.C.; Leitão, I.D.C.; Mariani, D.; Ota, V.A.; et al. Intra-Host Evolution during SARS-CoV-2 Prolonged Infection. Virus Evol. 2021, 7, veab078. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Quadeer, A.A.; Krishnan, P.; Ng, D.Y.M.; Chang, L.D.J.; Liu, G.Y.Z.; Cheng, S.M.S.; Lam, T.T.Y.; Peiris, M.; McKay, M.R.; et al. Within-Host Genetic Diversity of SARS-CoV-2 Lineages in Unvaccinated and Vaccinated Individuals. Nat. Commun. 2023, 14, 1793. [Google Scholar] [CrossRef] [PubMed]
- OSANG HEALTHCARE. GeneFinderTM COVID-19 Fast RealAmp Kit. Available online: https://www.osanghc.com/en/products_en/molecular-diagnosis/# (accessed on 24 November 2023).
- Brabaham Bioinformatics FastQC. A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 November 2023).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Castellano, S.; Cestari, F.; Faglioni, G.; Tenedini, E.; Marino, M.; Artuso, L.; Manfredini, R.; Luppi, M.; Trenti, T.; Tagliafico, E. Ivar, an Interpretation-oriented Tool to Manage the Update and Revision of Variant Annotation and Classification. Genes 2021, 12, 384. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. Available online: https://www.R-project.org (accessed on 24 November 2023).
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D. Ggplot2: Elegant Graphics for Data Analysis. Available online: https://ggplot2.tidyverse.org/ (accessed on 24 November 2023).
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so Different after All: A Comparison of Methods for Detecting Amino Acid Sites under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361.e6. [Google Scholar] [CrossRef]
- Fontanet, A.; Cauchemez, S. COVID-19 Herd Immunity: Where Are We? Nat. Rev. Immunol. 2020, 20, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Jena, D.; Ghosh, A.; Jha, A.; Prasad, P.; Raghav, S.K. Impact of Vaccination on SARS-CoV-2 Evolution and Immune Escape Variants. Vaccine 2024, 42, 126153. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Garcia-Segura, P.; Novau-Ferré, N.; Macip, G.; Martínez, R.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. The Mutational Landscape of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 9072. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5, 10–1128. [Google Scholar] [CrossRef]
- Fung, S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Gabriella Torcia, M.; Mattiuz, G.; Conticello, S.G. Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Hughes, M.A.K. More Effective Purifying Selection on RNA Viruses than in DNA Viruses. Gene 2007, 404, 117–125. [Google Scholar] [CrossRef]
- Ghafari, M.; Du Plessis, L.; Raghwani, J.; Bhatt, S.; Xu, B.; Pybus, O.G.; Katzourakis, A. Purifying Selection Determines the Short-Term Time Dependency of Evolutionary Rates in SARS-CoV-2 and PH1N1 Influenza. Mol. Biol. Evol. 2022, 39, msac009. [Google Scholar] [CrossRef]
- Holmes, E.C. Patterns of Intra- and Interhost Nonsynonymous Variation Reveal Strong Purifying Selection in Dengue Virus. J. Virol. 2003, 77, 11296–11298. [Google Scholar] [CrossRef]
- Riemersma, K.K.; Coffey, L.L. Chikungunya Virus Populations Experience Diversity- Dependent Attenuation and Purifying Intra-Vector Selection in Californian Aedes Aegypti Mosquitoes. PLOS Neglected Trop. Dis. 2019, 13, e0007853. [Google Scholar] [CrossRef]
- Krachmarova, E.; Petkov, P.; Lilkova, E.; Ilieva, N.; Rangelov, M.; Todorova, N.; Malinova, K.; Hristova, R.; Nacheva, G.; Gospodinov, A.; et al. Insights into the SARS-CoV-2 ORF6 Mechanism of Action. Int. J. Mol. Sci. 2023, 24, 11589. [Google Scholar] [CrossRef]
- Santos-Mendoza, T. The Envelope (E) Protein of SARS-CoV-2 as a Pharmacological Target. Viruses 2023, 15, 1000. [Google Scholar] [CrossRef]
- Bueno, S.M.; Abarca, K.; González, P.A.; Gálvez, N.M.S.; Soto, J.A.; Duarte, L.F.; Schultz, B.M.; Pacheco, G.A.; González, L.A.; Vázquez, Y.; et al. Safety and Immunogenicity of an Inactivated Severe Acute Respiratory Syndrome Coronavirus 2 Vaccine in a Subgroup of Healthy Adults in Chile. Clin. Infect. Dis. 2022, 75, E792–E804. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Gálvez, N.M.S.; Iturriaga, C.; Melo-González, F.; Soto, J.A.; Schultz, B.M.; Urzúa, M.; González, L.A.; Vázquez, Y.; Ríos, M.; et al. Immune Profile and Clinical Outcome of Breakthrough Cases after Vaccination with an Inactivated SARS-CoV-2 Vaccine. Front. Immunol. 2021, 12, 742914. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef]
- Trombetta, C.M.; Piccini, G.; Pierleoni, G.; Leonardi, M.; Dapporto, F.; Marchi, S.; Andreano, E.; Paciello, I.; Benincasa, L.; Lovreglio, P.; et al. Immune Response to SARS-CoV-2 Omicron Variant in Patients and Vaccinees Following Homologous and Heterologous Vaccinations. Commun. Biol. 2022, 5, 903. [Google Scholar] [CrossRef] [PubMed]
- Girl, P.; von Buttlar, H.; Mantel, E.; Antwerpen, M.H.; Wölfel, R.; Müller, K. Comparative Analysis of Vaccine-Induced Neutralizing Antibodies against the Alpha, Beta, Delta, and Omicron Variants of SARS-CoV-2. Vaccines 2024, 12, 515. [Google Scholar] [CrossRef] [PubMed]
- Feikin, D.R.; Higdon, M.M.; Abu-Raddad, L.J.; Andrews, N.; Araos, R.; Goldberg, Y.; Groome, M.J.; Huppert, A.; O’Brien, K.L.; Smith, P.G.; et al. Duration of Effectiveness of Vaccines against SARS-CoV-2 Infection and COVID-19 Disease: Results of a Systematic Review and Meta-Regression. Lancet 2022, 399, 924–944. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St. Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Planas, D.; Bruel, T.; Grzelak, L.; Guivel-Benhassine, F.; Staropoli, I.; Porrot, F.; Planchais, C.; Buchrieser, J.; Rajah, M.M.; Bishop, E.; et al. Sensitivity of Infectious SARS-CoV-2 B.1.1.7 and B.1.351 Variants to Neutralizing Antibodies. Nat. Med. 2021, 27, 917–924. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Unvaccinated | Vaccinated | |||
|---|---|---|---|---|
| Region | NS (%) | S (%) | NS (%) | S (%) |
| ORF1ab | 79 (58.5) | 74 (71.8) | 92 (54.8) | 96 (73.3) |
| S | 20 (14.8%) | 12 (11.7) | 27 (16.1) | 6 (4.6) |
| ORF3a | 9 (6.7) | 0 (0.0) | 13 (7.7) | 3 (2.3) |
| E | 0 (0.0) | 2 (1.9) | 0 (0.0) | 3 (2.3) |
| M | 0 (0.0) | 2 (1.9) | 3 (1.8) | 7 (5.3) |
| ORF6 | 3 (2.2) | 1 (1.0) | 3 (1.8) | 2 (1.5) |
| ORF7a | 2 (1.5) | 2 (1.9) | 4 (2.4) | 3 (2.3) |
| ORF7b | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| ORF8 | 6 (4.4) | 0 (0.0) | 5 (3.0) | 1 (0.8) |
| N | 16 (11.9) | 10 (9.8) | 20 (11.9) | 10 (7.6) |
| ORF10 | 0 (0.0) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
| TOTAL | 135 (100) | 103 (100) | 168 (100) | 131 (100) |
| Unvaccinated | Vaccinated | |||
|---|---|---|---|---|
| Locus | Positive Selection | Negative Selection | Positive Selection | Negative Selection |
| ORF1ab | NSP6 (106) | NSP3 (106, 681), NSP10 (82), NSP13 (495) | NSP3 (1303), NSP6 (107) | NSP2 (91,443), NSP3 (236, 394, 447, 662, 1092, 1121, 1742), NSP6 (76, 138), NSP10 (16), NSP13 (237, 356), NSP14 (302, 373), NSP15 (278), NSP16 (178) |
| S | 0 | 554, 995, 1065 | 0 | 0 |
| ORF3a | 0 | 0 | 0 | 43 |
| E | 0 | 0 | 0 | 8, 23 |
| M | 0 | 53 | 0 | 0 |
| ORF6 | 0 | 49 | 0 | 61 |
| ORF7a | 0 | 88 | 0 | 11 |
| ORF8 | 0 | 0 | 0 | 75 |
| N | 0 | 0 | 200 | 194, 363 |
| ORF10 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, B.d.C.; Banho, C.A.; Sacchetto, L.; Negri, A.; Vasilakis, N.; Nogueira, M.L. Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage. Viruses 2024, 16, 1524. https://doi.org/10.3390/v16101524
Marques BdC, Banho CA, Sacchetto L, Negri A, Vasilakis N, Nogueira ML. Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage. Viruses. 2024; 16(10):1524. https://doi.org/10.3390/v16101524
Chicago/Turabian StyleMarques, Beatriz de Carvalho, Cecília Artico Banho, Lívia Sacchetto, Andreia Negri, Nikos Vasilakis, and Maurício Lacerda Nogueira. 2024. "Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage" Viruses 16, no. 10: 1524. https://doi.org/10.3390/v16101524
APA StyleMarques, B. d. C., Banho, C. A., Sacchetto, L., Negri, A., Vasilakis, N., & Nogueira, M. L. (2024). Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage. Viruses, 16(10), 1524. https://doi.org/10.3390/v16101524