
Genomes of Alphanucleorhabdovirus Physostegiae Isolates from Two Different Cultivar Groups of Solanum melongena

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
3. Results
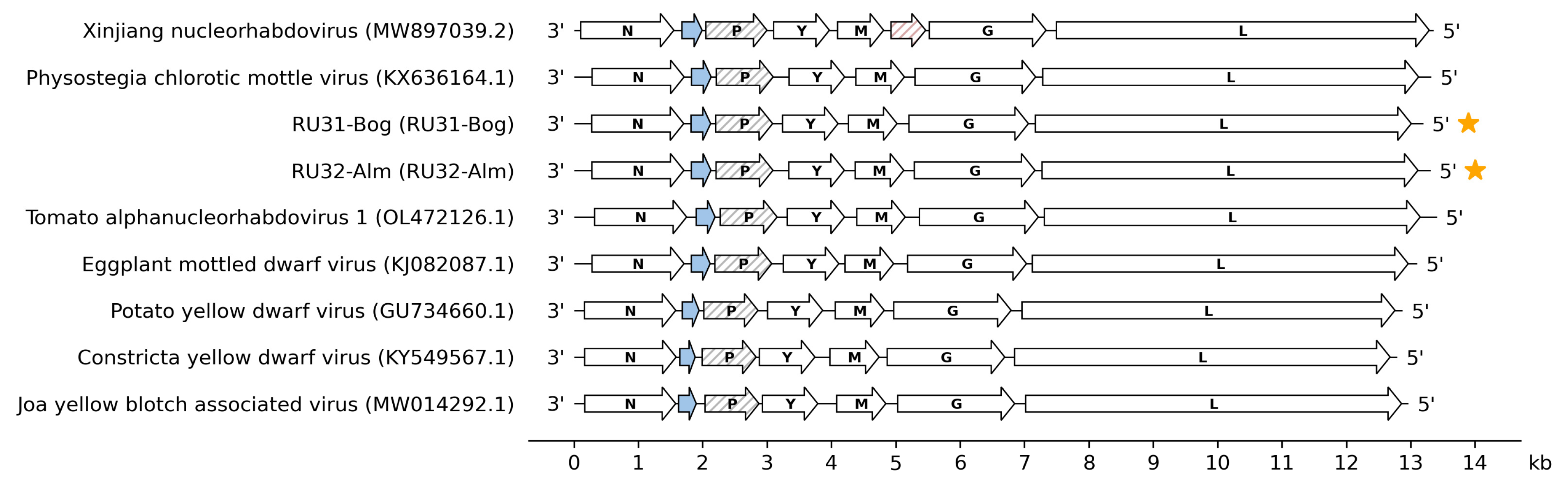
3.1. Genome Sequences of the Revealed Eggplant Rhabdoviruses
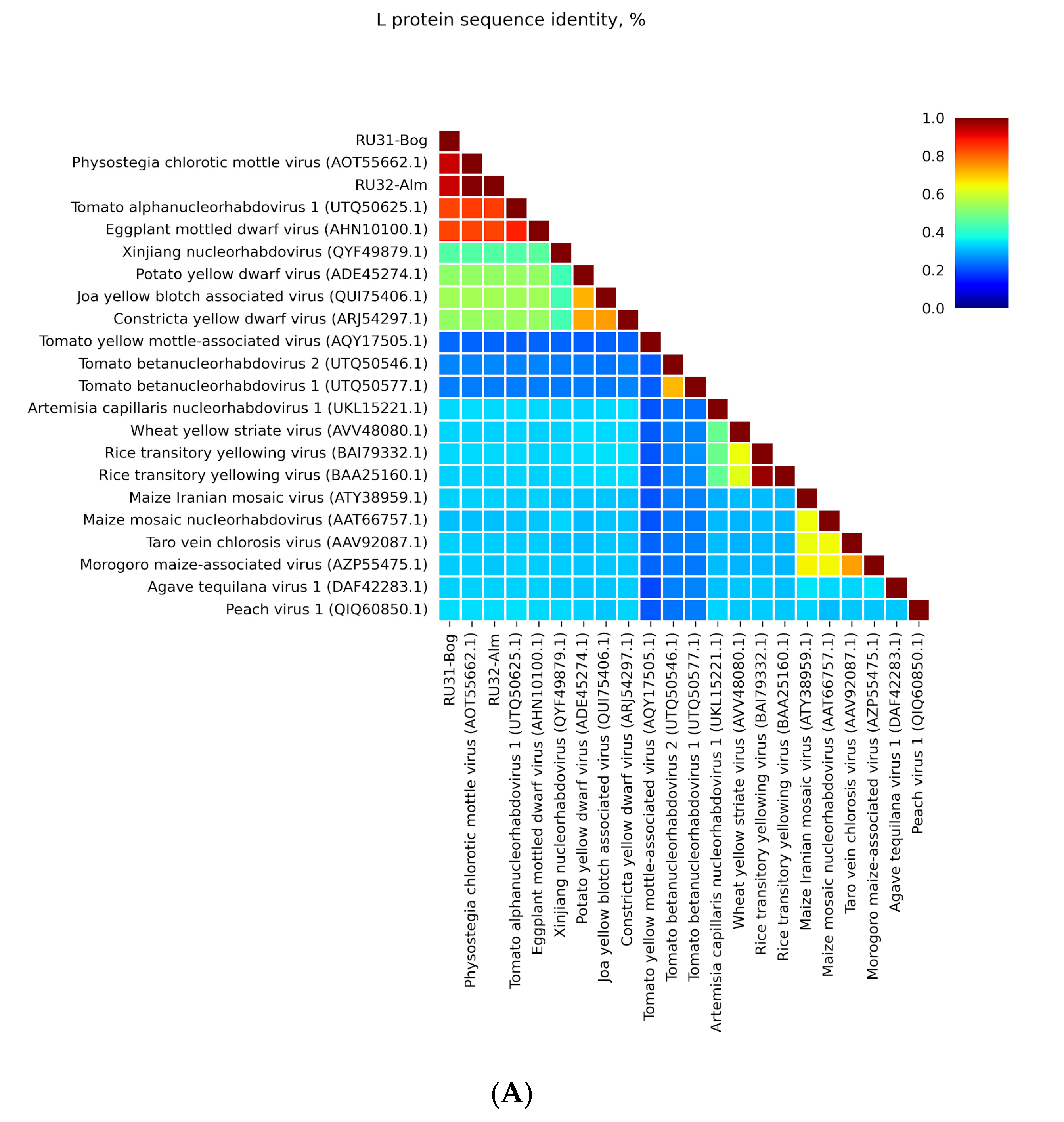
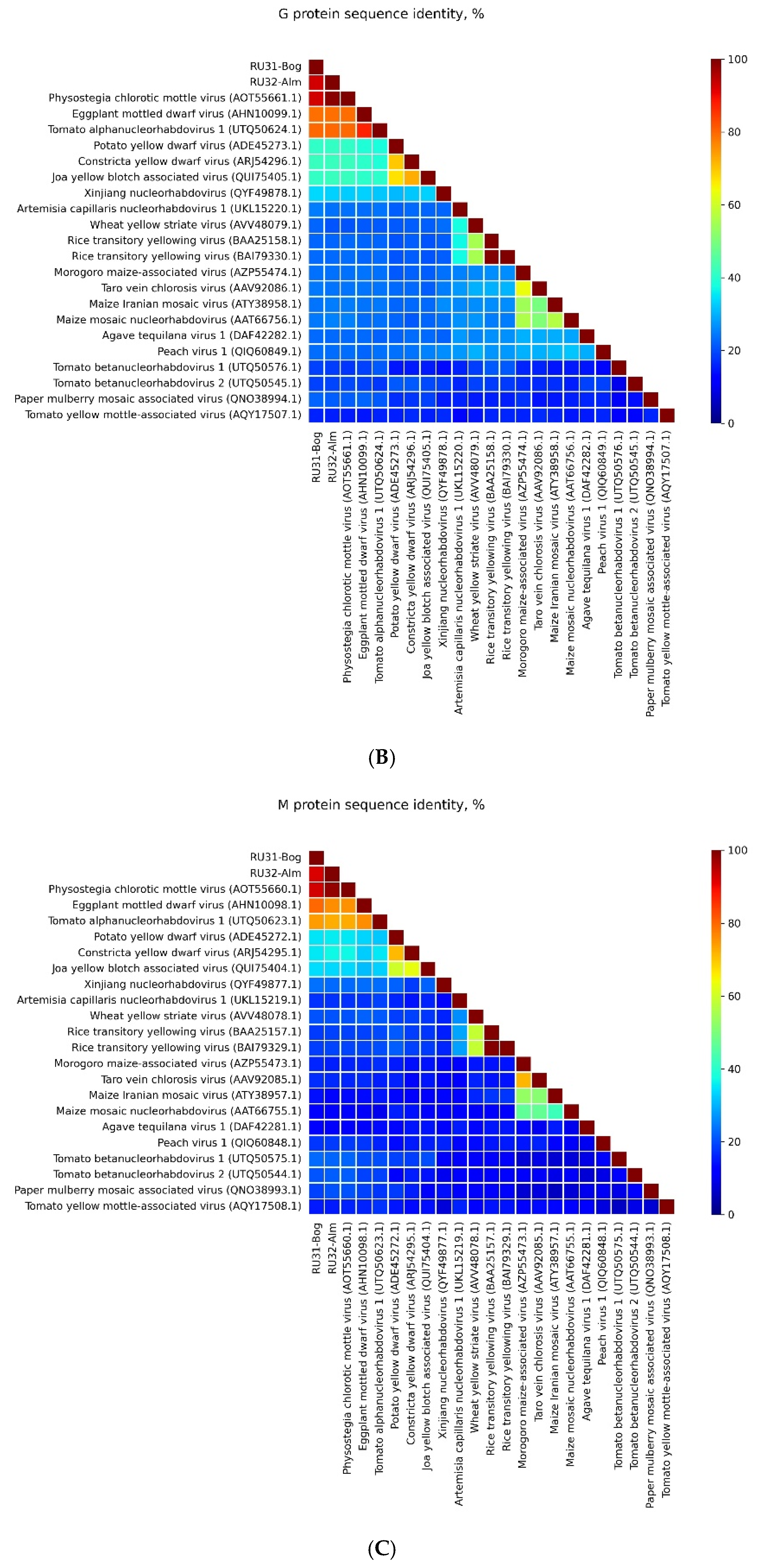
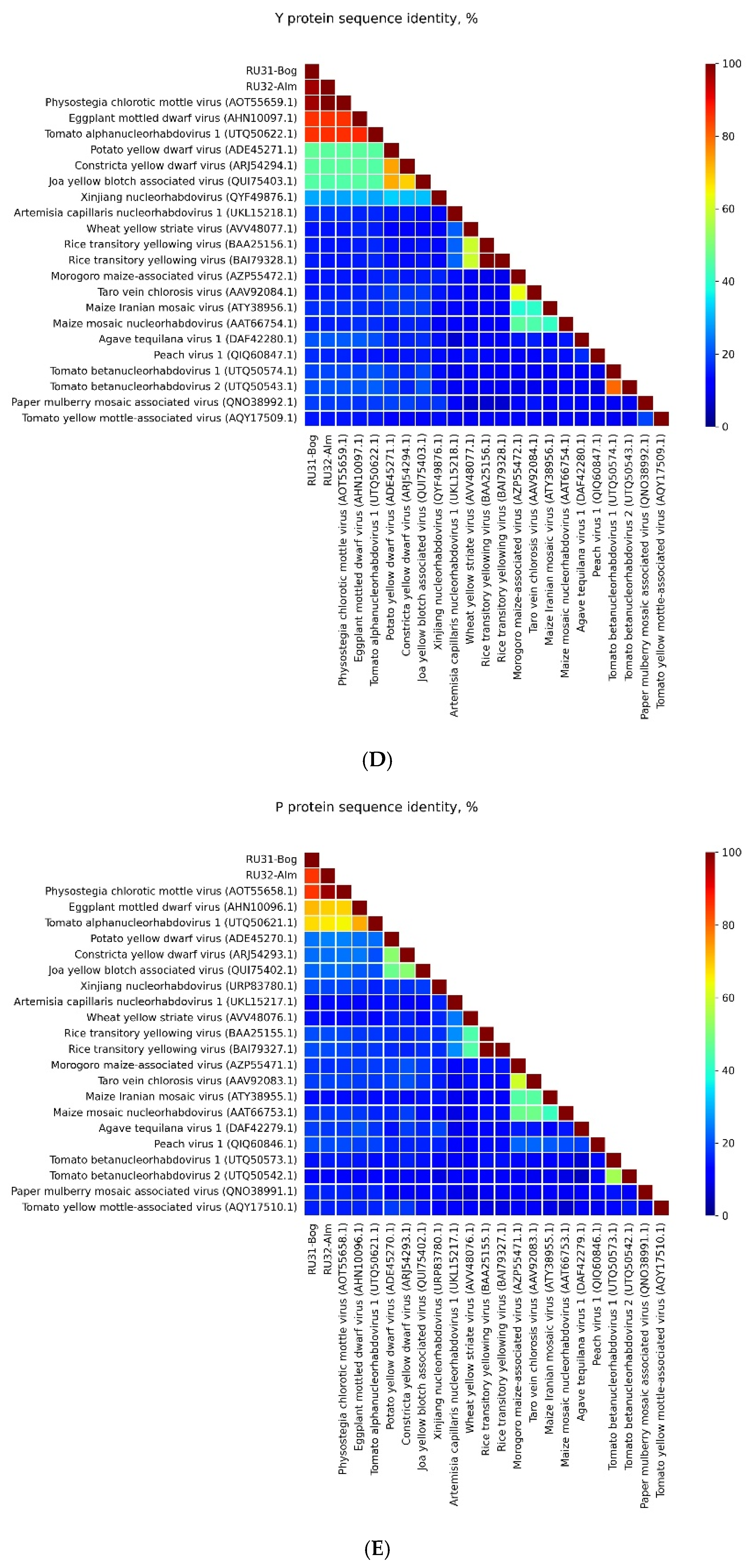
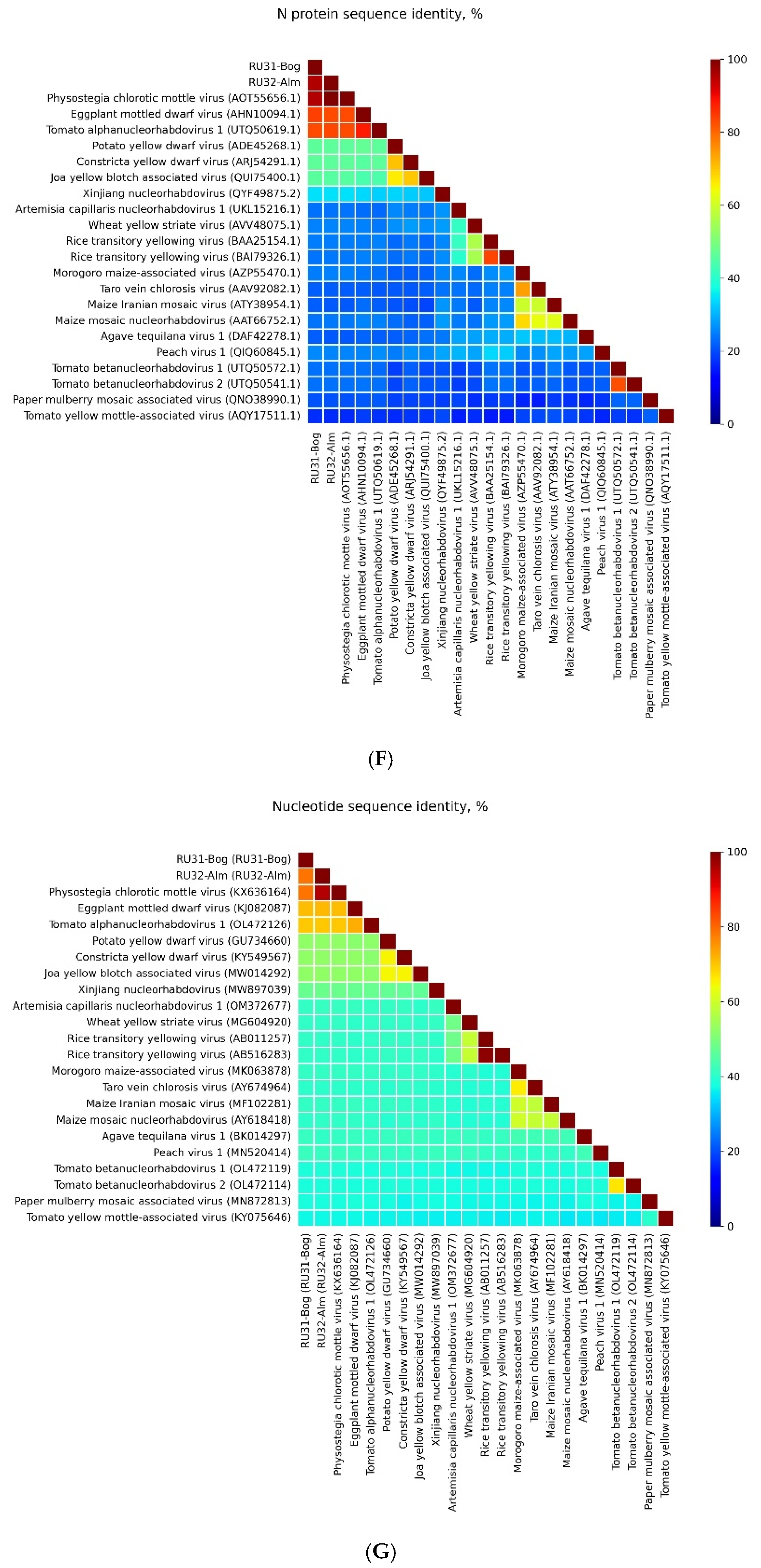
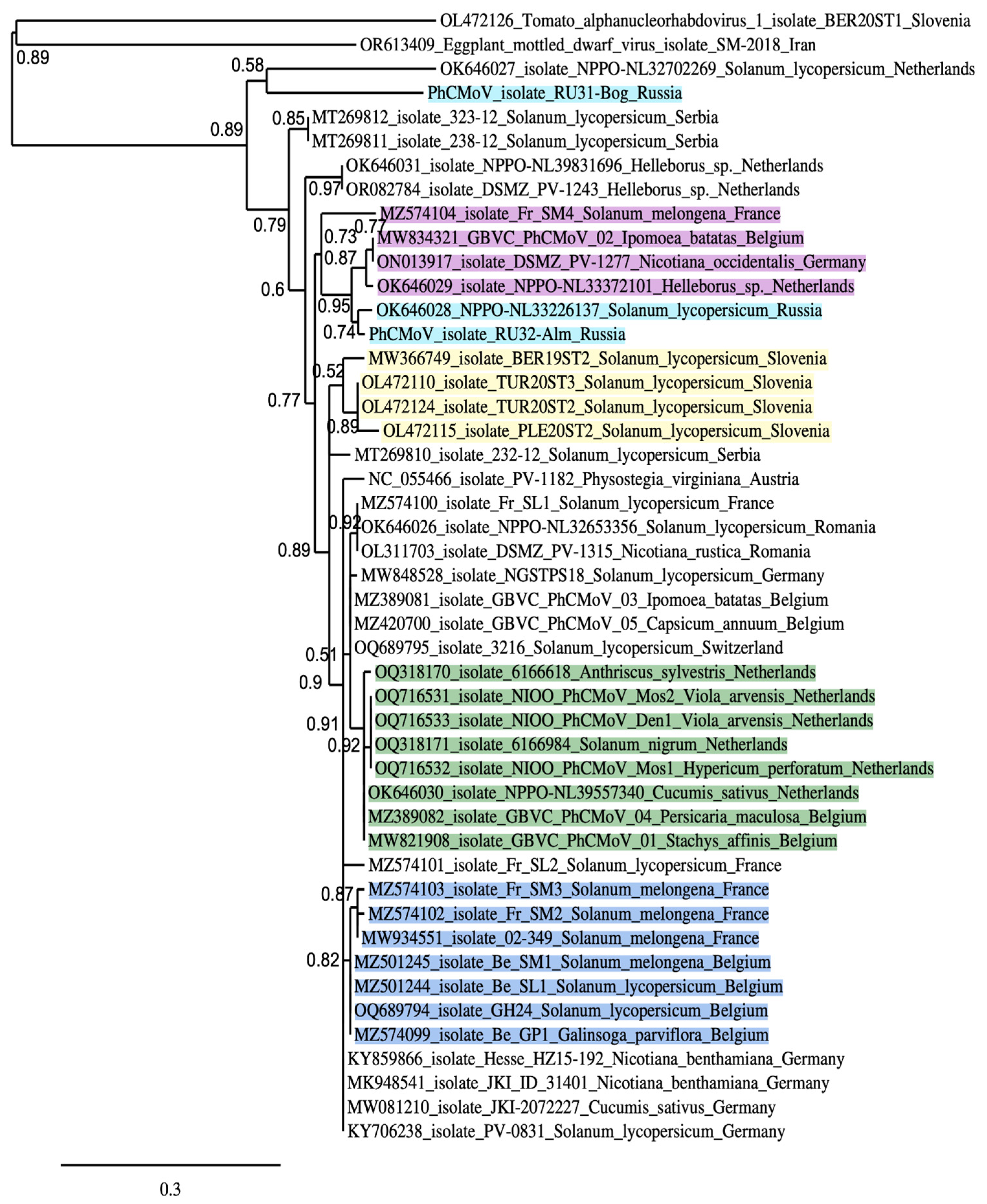
3.2. Placement of RU31-Bog and RU32-Alm in the Phylogenies of the Previously Sequenced PhCMoV Isolates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roossinck, M.J. Mechanisms of plant virus evolution. Annu. Rev. Phytopathol. 1997, 35, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Fraile, A.; Garcıa-Arenal, F. Evolution and emergence of plant viruses. Adv. Virus Res. 2014, 88, 161–191. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Holmes, E.C.; González-Candelas, F. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2004, 2, 279–288. [Google Scholar] [CrossRef] [PubMed]
- McLeish, M.J.; Fraile, A.; García-Arenal, F. Population Genomics of Plant Viruses: The Ecology and Evolution of Virus Emergence. Phytopathology 2021, 111, 32–39. [Google Scholar] [CrossRef]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of plant viruses and disease management: Relevance of genetic diversity and evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, Y.-F.; Deng, C.; Ma, Y.; Wang, Z.W.; Chen, X.H.; Xue, L.B. Comparative transcriptome analysis of eggplant (Solanum melongena L.) and Turkey berry (Solanum torvum Sw.): Phylogenomics and disease resistance analysis. BMC Genom. 2014, 15, 412. [Google Scholar] [CrossRef]
- Page, A.; Gibson, J.; Meyer, R.S.; Chapman, M.A. Eggplant domestication: Pervasive gene flow, feralisation and transcriptomic divergence. Mol. Biol. Evol. 2019, 36, 1359–1372. [Google Scholar] [CrossRef]
- Garcia-Estrada, R.S.; Cruz-Lachica, I.; Osuna-Garcia, L.A.; Marquez, I. First Report of Eggplant Fruit Rot Caused by Phytophthora nicotianae Breda de Haan in Mexico. Plant Dis. 2021, 105, 513. [Google Scholar] [CrossRef] [PubMed]
- Scholthof, K.-B.G. The Past Is Present: Coevolution of Viruses and Host Resistance within Geographic Centers of Plant Diversity. Annu. Rev. Phytopathol. 2023, 61, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Gitaitis, R.; Dutta, B. Characterization of Pseudomonas capsici strains from pepper and tomato. Front. Microbiol. 2023, 14, 1267395. [Google Scholar] [CrossRef]
- Temple, C.; Blouin, A.G.; De Jonghe, K.; Foucart, Y.; Botermans, M.; Westenberg, M.; Schoen, R.; Gentit, P.; Visage, M.; Verdin, E.; et al. Biological and Genetic Characterization of Physostegia Chlorotic Mottle Virus in Europe Based on Host Range, Location, and Time. Plant Dis. 2022, 106, 2797–2807. [Google Scholar] [CrossRef] [PubMed]
- Temple, C.; Blouin, A.G.; Boezen, D.; Botermans, M.; Durant, L.; De Jonghe, K.; de Koning, P.; Goedefroit, T.; Minet, L.; Steyer, S.; et al. Biological Characterization of Physostegia Chlorotic Mottle Virus, an Emergent Virus Infecting Vegetables in Diversified Production Systems. Phytopathology 2024, 114, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Alfaro-Fernández, A.; Taengua, R.; Font-San-Ambrosio, I.; Sanahuja-Edo, E.; Peiró, R.; Galipienso, L.; Rubio, L. Genetic Variation and Evolutionary Analysis of Eggplant Mottled Dwarf Virus Isolates from Spain. Plants 2024, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Pappi, P.G.; Dovas, C.I.; Efthimiou, K.E.; Maliogka, V.I.; Katis, N.I. A novel strategy for the determination of a rhabdovirus genome and its application to sequencing of Eggplant mottled dwarf virus. Virus Genes 2013, 47, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus accessory genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Dietzgen, R.G.; Debat, H. Illuminating the Plant Rhabdovirus Landscape through Metatranscriptomics Data. Viruses 2021, 13, 1304. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Bejerman, N.E.; Goodin, M.M.; Higgins, C.M.; Huot, O.B.; Kondo, H.; Martin, K.M.; Whitfield, A.E. Diversity and epidemiology of plant rhabdoviruses. Virus Res. 2020, 281, 197942. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Bejerman, N.E.; Mei, Y.; Jee, C.L.J.; Chabi-Jesus, C.; Freitas-Astúa, J.; Veras, S.M.; Kitajima, E.W. Joá yellow blotch-associated virus, a new alphanucleorhabdovirus from a wild solanaceous plant in Brazil. Arch. Virol. 2021, 166, 1615–1622. [Google Scholar] [CrossRef]
- Jang, C.; Wang, R.; Wells, J.; Leon, F.; Farman, M.; Hammond, J.; Goodin, M.M. Genome sequence variation in the constricta strain dramatically alters the protein interaction and localization map of Potato yellow dwarf virus. J. Gen. Virol. 2017, 98, 1526–1536. [Google Scholar] [CrossRef]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef]
- Pappi, P.G.; Maliogka, V.I.; Amoutzias, G.D.; Katis, N.I. Genetic variation of eggplant mottled dwarf virus from annual and perennial plant hosts. Arch. Virol. 2016, 161, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Boonham, N.; Kreuze, J.; Winter, S.; van der Vlugt, R.; Bergervoet, J.; Tomlinson, J.; Mumford, R. Methods in virus diagnostics: From ELISA to next generation sequencing. Virus Res. 2014, 186, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Debat, H.; Dietzgen, R.G. The Plant Negative-Sense RNA Virosphere: Virus Discovery Through New Eyes. Front. Microbiol. 2020, 11, 588427. [Google Scholar] [CrossRef] [PubMed]
- German, T.L.; Lorenzen, M.D.; Grubbs, N.; Whitfield, A.E. New Technologies for Studying Negative-Strand RNA Viruses in Plant and Arthropod Hosts. Mol. Plant-Microbe Interact. 2020, 33, 382–393. [Google Scholar] [CrossRef]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Roossinck, M.J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef] [PubMed]
- Meleshko, D.; Hajirasouliha, I.; Korobeynikov, A. coronaSPAdes: From biosynthetic gene clusters to RNA viral assemblies. Bioinformatics 2021, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Menzel, W.; Richert-Poggeler, K.; Winter, S.; Knierim, D. Characterization of a nucleorhabdovirus from Physostegia. Acta Hortic. 2018, 1193, 29–38. [Google Scholar] [CrossRef]
- Vučurović, A.; Kutnjak, D.; Mehle, N.; Stanković, I.; Pecman, A.; Bulajić, A.; Krstić, B.; Ravnikar, M. Detection of Four New Tomato Viruses in Serbia Using Post Hoc High-Throughput Sequencing Analysis of Samples from a Large-Scale Field Survey. Plant Dis. 2021, 105, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Gaafar, Y.Z.A.; Abdelgalil, M.A.M.; Knierim, D.; Richert-Poggeler, K.R.; Menzel, W.; Winter, S.; Ziebell, H. First report of physostegia chlorotic mottle virus on tomato (Solanum lycopersicum) in Germany. Plant Dis. 2018, 102, 255. [Google Scholar] [CrossRef]
- Parrella, G.; Greco, B. Sequence variation of block III segment identifies three distinct lineages within Eggplant mottled dwarf virus isolates from Italy, Spain and Greece. Acta Virol. 2016, 60, 100–105. [Google Scholar] [CrossRef]
- Yang, X.; Chen, B.; Zhang, T.; Li, Z.; Xu, C.; Zhou, G. Geographic distribution and genetic diversity of rice stripe mosaic virus in Southern China. Front. Microbiol. 2018, 9, 3068. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location of GGG Sequence in Leader and Intercistronic Regions of Genomic RNA | Nucleotide Position of Central G Residue in RU31-Bog * | Nucleotide Position of Central G Residue in RU32-Alm ** |
|---|---|---|
| Leader/N gene junction | 13,001 | 13,109 |
| N gene/X gene | 11,388 | 11,493 |
| X gene/P gene | 11,017 | 11,122 |
| P gene/Y (MP) gene | 9968 | 9981 |
| Y (MP) gene/M gene | 8943 | 8948 |
| M gene/G gene | 8029 | 8037 |
| G gene/L gene | 6038 | 6043 |
| PhCMoV Strains * | E-Value | Percentage of Identity | NCBI Accession |
|---|---|---|---|
| isolate GBVC_PhCMoV_02_IB | 0.0 | 97.90% | MW834321 |
| isolate DSMZ PV-1277 | 0.0 | 97.86% | ON013917 |
| isolate NPPO-NL33372101 | 0.0 | 97.89% | OK646029 |
| isolate NPPO-NL33226137 | 0.0 | 96.71% | OK646028 |
| isolate Fr_SM4 | 0.0 | 96.22% | MZ574104 |
| isolate PV-1182 | 0.0 | 95.42% | NC055466 |
| isolate Be_GP1 | 0.0 | 95.40% | MZ574099 |
| isolate 3216 | 0.0 | 95.40% | OQ689795 |
| isolate 232–12 | 0.0 | 95.36% | MT269810 |
| PhCMoV Strains * | E-Value | Percentage of Identity | NCBI Accession |
|---|---|---|---|
| isolate BER19ST2 | 0.0 | 100.00% | MW366749 |
| isolate TUR20ST2 | 0.0 | 97.67% | OL472124 |
| isolate TUR20ST3 | 0.0 | 97.67% | OL472110 |
| isolate PLE20ST2 | 0.0 | 97.63% | OL472115 |
| isolate Be_GP1 | 0.0 | 96.41% | MZ574099 |
| isolate GH24 | 0.0 | 96.43% | OQ689794 |
| isolate 3216 | 0.0 | 95.40% | OQ689795 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gryzunov, N.; Morozov, S.Y.; Suprunova, T.; Samarskaya, V.; Spechenkova, N.; Yakunina, S.; Kalinina, N.O.; Taliansky, M. Genomes of Alphanucleorhabdovirus Physostegiae Isolates from Two Different Cultivar Groups of Solanum melongena. Viruses 2024, 16, 1538. https://doi.org/10.3390/v16101538
Gryzunov N, Morozov SY, Suprunova T, Samarskaya V, Spechenkova N, Yakunina S, Kalinina NO, Taliansky M. Genomes of Alphanucleorhabdovirus Physostegiae Isolates from Two Different Cultivar Groups of Solanum melongena. Viruses. 2024; 16(10):1538. https://doi.org/10.3390/v16101538
Chicago/Turabian StyleGryzunov, Nikita, Sergey Yu. Morozov, Tatiana Suprunova, Viktoriya Samarskaya, Nadezhda Spechenkova, Sofiya Yakunina, Natalia O. Kalinina, and Michael Taliansky. 2024. "Genomes of Alphanucleorhabdovirus Physostegiae Isolates from Two Different Cultivar Groups of Solanum melongena" Viruses 16, no. 10: 1538. https://doi.org/10.3390/v16101538
APA StyleGryzunov, N., Morozov, S. Y., Suprunova, T., Samarskaya, V., Spechenkova, N., Yakunina, S., Kalinina, N. O., & Taliansky, M. (2024). Genomes of Alphanucleorhabdovirus Physostegiae Isolates from Two Different Cultivar Groups of Solanum melongena. Viruses, 16(10), 1538. https://doi.org/10.3390/v16101538