
Full Genome Characterization of the First Oropouche Virus Isolate Imported in Europe from Cuba
 ,
,  , , ,
, , ,  , ,
, ,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Isolation
2.2. RNA Extraction, Library Preparation and Sequencing
2.3. Bioinformatic Analysis
2.3.1. Reads Processing, Assembly and Consensus
2.3.2. Variant Calling and Annotation
2.3.3. GenBank Data Download
2.3.4. Phylogenetic Analysis and Genetic Variability
3. Results
3.1. Quality Control (QC) Analyses, Read Alignment and Consensus Generation
3.2. Variant Calling and Annotation
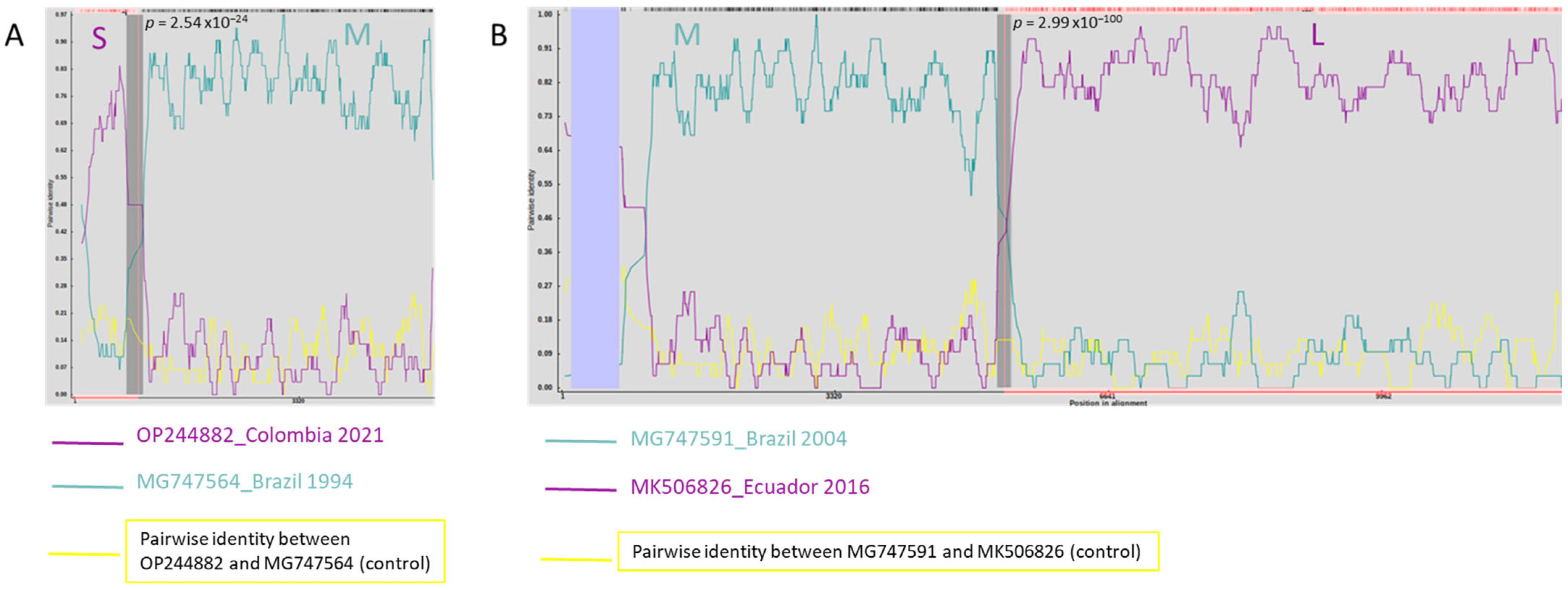
3.3. Phylogenetic Analysis and Genetic Variability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ministry of Public Health of Cuba. Information Note from the Ministry of Public Health 2024. Available online: https://salud.msp.gob.cu/nota-informativa-del-ministerio-de-salud-publica-8/ (accessed on 29 September 2024).
- Wesselmann, K.M.; Postigo-Hidalgo, I.; Pezzi, L.; De Oliveira-Filho, E.F.; Fischer, C.; De Lamballerie, X.; Drexler, J.F. Emergence of Oropouche Fever in Latin America: A Narrative Review. Lancet Infect. Dis. 2024, 24, e439–e452. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, H.; Bozidis, P.; Franks, A.; Papadopoulou, C. Oropouche Fever: A Review. Viruses 2018, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Pan American Health Organization/World Health Organization (PAHO/WHO) Epidemiological Alert: Oropouche in the Region of the Americas: Vertical Transmission Event under Investigation in Brazil. 2024. Available online: https://www.paho.org/en/documents/epidemiological-alert-oropouche-region-americas-vertical-transmission-event-under (accessed on 29 September 2024).
- Republica de Cuba, Ministerio de Salud Pública Informan Autoridades Del Minsap Sobre Situación Epidemiológica En Cuba, 24 June 2024. Available online: https://salud.msp.gob.cu/informan-autoridades-del-minsap-sobre-situacion-epidemiologica-en-cuba/ (accessed on 29 September 2024).
- Hughes, H.R.; Adkins, S.; Alkhovskiy, S.; Beer, M.; Blair, C.; Calisher, C.H.; Drebot, M.; Lambert, A.J.; De Souza, W.M.; Marklewitz, M.; et al. ICTV Virus Taxonomy Profile: Peribunyaviridae. J. Gen. Virol. 2020, 101, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Ladner, J.T.; Savji, N.; Lofts, L.; Travassos Da Rosa, A.; Wiley, M.R.; Gestole, M.C.; Rosen, G.E.; Guzman, H.; Vasconcelos, P.F.C.; Nunes, M.R.T.; et al. Genomic and Phylogenetic Characterization of Viruses Included in the Manzanilla and Oropouche Species Complexes of the Genus Orthobunyavirus, Family Bunyaviridae. J. Gen. Virol. 2014, 95, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.V.; Barrett, A.D.; Saeed, M.F.; Watts, D.M.; Russell, K.; Guevara, C.; Ampuero, J.S.; Suarez, L.; Cespedes, M.; Montgomery, J.M.; et al. Iquitos Virus: A Novel Reassortant Orthobunyavirus Associated with Human Illness in Peru. PLoS Negl. Trop. Dis. 2011, 5, e1315. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and Pandemic Potential of Swine-Origin H1N1 Influenza Virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef]
- Navarro, J.-C.; Giambalvo, D.; Hernandez, R.; Auguste, A.J.; Tesh, R.B.; Weaver, S.C.; Montañez, H.; Liria, J.; Lima, A.; Travassos Da Rosa, J.F.S.; et al. Isolation of Madre de Dios Virus (Orthobunyavirus; Bunyaviridae), an Oropouche Virus Species Reassortant, from a Monkey in Venezuela. Am. Soc. Trop. Med. Hyg. 2016, 95, 328–338. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; De Souza, W.M.; Savji, N.; Figueiredo, M.L.; Cardoso, J.F.; Da Silva, S.P.; Da Silva De Lima, C.P.; Vasconcelos, H.B.; Rodrigues, S.G.; Ian Lipkin, W.; et al. Oropouche Orthobunyavirus: Genetic Characterization of Full-Length Genomes and Development of Molecular Methods to Discriminate Natural Reassortments. Infect. Genet. Evol. 2019, 68, 16–22. [Google Scholar] [CrossRef]
- Vasconcelos, H.B.; Nunes, M.R.T.; Casseb, L.M.N.; Carvalho, V.L.; Pinto Da Silva, E.V.; Silva, M.; Casseb, S.M.M.; Vasconcelos, P.F.C. Molecular Epidemiology of Oropouche Virus, Brazil. Emerg. Infect. Dis. 2011, 17, 800–806. [Google Scholar] [CrossRef]
- Castilletti, C.; Mori, A.; Matucci, A.; Ronzoni, N.; Van Duffel, L.; Rossini, G.; Sponga, P.; D’Errico, M.L.; Rodari, P.; Cristini, F.; et al. Oropouche Fever Cases Diagnosed in Italy in Two Epidemiologically Non-Related Travellers from Cuba, Late May to Early June 2024. Eurosurveillance 2024, 29, 2400362. [Google Scholar] [CrossRef] [PubMed]
- Acrani, G.O.; Tilston-Lunel, N.L.; Spiegel, M.; Weidmann, M.; Dilcher, M.; Andrade Da Silva, D.E.; Nunes, M.R.T.; Elliott, R.M. Establishment of a Minigenome System for Oropouche Virus Reveals the S Genome Segment to Be Significantly Longer than Reported Previously. J. Gen. Virol. 2015, 96, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.R.R.; Dutra, J.V.R.; de Carvalho, A.H.B.; Reis, C.R.; Rios, J.S.H.; Ribeiro, M.d.O.; Arruda, M.B.; Alvarez, P.; Souza, R.P.; Voloch, C.; et al. Oropouche Virus Genomic Surveillance in Brazil. Lancet Infect. Dis. 2024. [Google Scholar] [CrossRef]
- Moreira, H.M.; Sgorlon, G.; Queiroz, J.A.S.; Roca, T.P.; Ribeiro, J.; Teixeira, K.S.; Passos-Silva, A.M.; Araújo, A.; Gasparelo, N.W.F.; Dos Santos, A.D.O.; et al. Outbreak of Oropouche Virus in Frontier Regions in Western Amazon. Microbiol. Spectr. 2024, 12, e01629-23. [Google Scholar] [CrossRef]
- Naveca, F.G.; De Almeida, T.A.P.; Souza, V.; Nascimento, V.; Silva, D.; Nascimento, F.; Mejía, M.; De Oliveira, Y.S.; Rocha, L.; Xavier, N.; et al. Human Outbreaks of a Novel Reassortant Oropouche Virus in the Brazilian Amazon Region. Nat. Med. 2024. [Google Scholar] [CrossRef]
- Iani, F.C.D.M.; Mota Pereira, F.; De Oliveira, E.C.; Nascimento Rodrigues, J.T.; Hoffmann Machado, M.; Fonseca, V.; Ribeiro Adelino, T.E.; Rocha Guimarães, N.; Ribeiro Tomé, L.M.; Astete Gómez, M.K.; et al. Rapid Viral Expansion Beyond the Amazon Basin: Increased Epidemic Activity of Oropouche Virus Across the Americas. MedRxiv 2024. [Google Scholar] [CrossRef]
- Gutierrez, B.; Wise, E.L.; Pullan, S.T.; Logue, C.H.; Bowden, T.A.; Escalera-Zamudio, M.; Trueba, G.; Nunes, M.R.T.; Faria, N.R.; Pybus, O.G. Evolutionary Dynamics of Oropouche Virus in South America. J. Virol. 2020, 94, e01127-19. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Castilletti, C.; Gobbi, F.G. Potential Risks of Oropouche Virus Importation into Europe. J. Travel Med. 2024, taae109. [Google Scholar] [CrossRef] [PubMed]
- Gaillet, M.; Pichard, C.; Restrepo, J.; Lavergne, A.; Perez, L.; Enfissi, A.; Abboud, P.; Lambert, Y.; Ma, L.; Monot, M.; et al. Outbreak of Oropouche Virus in French Guiana. Emerg. Infect. Dis. 2021, 27, 2711–2714. [Google Scholar] [CrossRef] [PubMed]
- Ciuoderis, K.A.; Berg, M.G.; Perez, L.J.; Hadji, A.; Perez-Restrepo, L.S.; Aristizabal, L.C.; Forberg, K.; Yamaguchi, J.; Cardona, A.; Weiss, S.; et al. Oropouche Virus as an Emerging Cause of Acute Febrile Illness in Colombia. Emerg. Microbes Infect. 2022, 11, 2645–2657. [Google Scholar] [CrossRef]
- Wise, E.L.; Márquez, S.; Mellors, J.; Paz, V.; Atkinson, B.; Gutierrez, B.; Zapata, S.; Coloma, J.; Pybus, O.G.; Jackson, S.K.; et al. Oropouche Virus Cases Identified in Ecuador Using an Optimised qRT-PCR Informed by Metagenomic Sequencing. PLoS Negl. Trop. Dis. 2020, 14, e0007897. [Google Scholar] [CrossRef] [PubMed]
- Files, M.A.; Hansen, C.A.; Herrera, V.C.; Schindewolf, C.; Barrett, A.D.T.; Beasley, D.W.C.; Bourne, N.; Milligan, G.N. Baseline Mapping of Oropouche Virology, Epidemiology, Therapeutics, and Vaccine Research and Development. Npj Vaccines 2022, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Scachetti, G.C.; Forato, J.; Claro, I.M.; Hua, X.; Salgado, B.B.; Vieira, A.; Simeoni, C.L.; Barbosa, A.R.C.; Rosa, I.L.; De Souza, G.F.; et al. Reemergence of Oropouche Virus between 2023 and 2024 in Brazil. MedRxiv 2024. [Google Scholar] [CrossRef]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Annu. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Accession ID | Total Number of Sequenced Fragments | Fragments Mapped on OROV (n) | Coverage (%) | Mean Depth Coverage (X) | 10× (%) | 30× (%) | 1000× (%) | |
|---|---|---|---|---|---|---|---|---|
| S | PP952117 | 88,041,642 | 5632 | 99.27 | 606 | 99.16 | 98.95 | 19.41 |
| M | PP952118 | 88,041,642 | 34,481 | 99.73 | 991 | 99.15 | 92.26 | 38.15 |
| L | PP952119 | 88,041,642 | 15,899 | 99.31 | 246 | 97.03 | 93.03 | 4.27 |
| Accession ID | Assembly Length | Coverage (%) | # SNVs | # Dels. | # Ins. | # Missense | # Synonymous | |
|---|---|---|---|---|---|---|---|---|
| S | PP952117 | 951 | 99.27 | 38 | 1 | 0 | 2 | 34 |
| M | PP952118 | 4400 | 99.73 | 165 | 0 | 0 | 23 | 138 |
| L | PP952119 | 7700 | 99.32 | 727 | 0 | 1 | 54 | 637 |
| Gen I | Gen II | Gen III | Gen IV | Mean Intragroup Divergence | |
|---|---|---|---|---|---|
| Gen I | - | 0.024 | |||
| Gen II | 5.0 ± 0.6 | - | 0.018 | ||
| Gen III | 5.9 ± 0.7 | 3.9 ± 0.6 | - | 0.007 | |
| Gen IV | 6.2 ± 0.8 | 5.6 ± 0.7 | 6.4 ± 0.9 | - | 0.004 |
| OROV_SCDC_2024 | 5.1 ± 0.6 | 3.4 ± 0.5 | 4.8 ± 0.7 | 6.0 ± 0.8 | 0.010 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deiana, M.; Malagò, S.; Mori, A.; Accordini, S.; Matucci, A.; Passarelli Mantovani, R.; Gianesini, N.; Huits, R.; Piubelli, C.; Gobbi, F.G.; et al. Full Genome Characterization of the First Oropouche Virus Isolate Imported in Europe from Cuba. Viruses 2024, 16, 1586. https://doi.org/10.3390/v16101586
Deiana M, Malagò S, Mori A, Accordini S, Matucci A, Passarelli Mantovani R, Gianesini N, Huits R, Piubelli C, Gobbi FG, et al. Full Genome Characterization of the First Oropouche Virus Isolate Imported in Europe from Cuba. Viruses. 2024; 16(10):1586. https://doi.org/10.3390/v16101586
Chicago/Turabian StyleDeiana, Michela, Simone Malagò, Antonio Mori, Silvia Accordini, Andrea Matucci, Rebeca Passarelli Mantovani, Natasha Gianesini, Ralph Huits, Chiara Piubelli, Federico Giovanni Gobbi, and et al. 2024. "Full Genome Characterization of the First Oropouche Virus Isolate Imported in Europe from Cuba" Viruses 16, no. 10: 1586. https://doi.org/10.3390/v16101586
APA StyleDeiana, M., Malagò, S., Mori, A., Accordini, S., Matucci, A., Passarelli Mantovani, R., Gianesini, N., Huits, R., Piubelli, C., Gobbi, F. G., Capobianchi, M. R., & Castilletti, C. (2024). Full Genome Characterization of the First Oropouche Virus Isolate Imported in Europe from Cuba. Viruses, 16(10), 1586. https://doi.org/10.3390/v16101586