
Molecular Characterization and Genomic Surveillance of SARS-CoV-2 Lineages in Central India

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition
2.2. RNA Extraction and Real-Time Reverse Transcriptase PCR (RT-PCR)
2.3. Library Preparation and Whole-Genome Sequencing
2.4. Variant Calling and Lineage Classification
2.5. Comparison with Global Strains

{kind=link}
{kind=link}
{kind=link}
| Sample-ID | Age (In Years) | Gender | District of Residence | Travel History/Contact | Date of Sample Collection | Sample Type | Symptoms |
|---|---|---|---|---|---|---|---|
| ICMR-NIRTH-S1 | 46 | M | JABALPUR | NA | 27 March 2020 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S2 | 48 | M | JABALPUR | NA | 27 March 2020 | Oropharyngeal swab | fever |
| ICMR-NIRTH-S3 | 24 | M | JABALPUR | Germany | 20 March 2020 | Nasopharyngeal swab | fever cough |
| ICMR-NIRTH-S4 | 59 | M | JABALPUR | Dubai, UAE | 20 March 2020 | Nasopharyngeal swab | fever cough |
| ICMR-NIRTH-S5 | 45 | F | JABALPUR | Dubai, UAE | 20 March 2020 | Nasopharyngeal swab | cough sore throat |
| ICMR-NIRTH-S6 | 22 | F | JABALPUR | Dubai, UAE | 20 March 2020 | Nasopharyngeal swab | cough |
| ICMR-NIRTH-S7 | 53 | M | JABALPUR | Dubai (contact of ICMR-NIRTH-S4) | 21 March 2020 | Nasopharyngeal swab | cough |
| ICMR-NIRTH-S8 | 40 | M | JABALPUR | NA | 22 March 2020 | Nasopharyngeal swab | cough body ache |
| ICMR-NIRTH-S11 | 33 | M | CHHINDWARA | Indore travel history | 1 April 2020 | Nasopharyngeal swab | fever cough breathlessness sore throat body ach |
| ICMR-NIRTH-S12 | 26 | F | CHHINDWARA | Delhi travel history | 7 April 2020 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S13 | 27 | M | CHHINDWARA | Contact of ICMR-NIRTH-S12 | 7 April 2020 | Nasopharyngeal swab | cough sore throat |
| ICMR-NIRTH-S14 | 61 | M | JABALPUR | Delhi travel history | 8 April 2020 | Nasopharyngeal swab | Fever cough breathlessness body ach sputum |
| ICMR-NIRTH-S15 | 30 | M | INDORE | Indore travel history | 10 April 2020 | Nasopharyngeal swab | fever cough sore throat |
| ICMR-NIRTH-S16 | 22 | M | INDORE | Indore travel history | 10 April 2020 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S17 | 24 | M | SATNA | Indore travel history | 10 April 2020 | Nasopharyngeal swab | cough sore throat |
| ICMR-NIRTH-S18 | 70 | M | JABALPUR | NA | 11 April 2020 | Nasopharyngeal swab | fever cough sore throat |
| ICMR-NIRTH-S19 | 50 | M | JABALPUR | Contact of ICMR-NIRTH-S18 | 14 April 2020 | Nasopharyngeal swab | fever sore throat |
| ICMR-NIRTH-S22 | 44 | F | JABALPUR | Contact of ICMR-NIRTH-S19 | 17 April 2020 | Throat swab | cough sore throat |
| ICMR-NIRTH-S23 | 35 | M | BALAGHAT | NA | 27 July 2020 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S24 | 28 | F | BALAGHAT | NA | 16 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S25 | 28 | M | BALAGHAT | NA | 16 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S26 | 24 | M | BALAGHAT | NA | 17 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S27 | 24 | M | BALAGHAT | NA | 18 April 2021 | Throat swab | Symptomatic |
| ICMR-NIRTH-S28 | 38 | M | BALAGHAT | NA | 19 April 2021 | Throat swab | Symptomatic |
| ICMR-NIRTH-S29 | 25 | M | BALAGHAT | NA | 19 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S30 | 19 | M | BALAGHAT | NA | 20 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S31 | 14 | M | BALAGHAT | NA | 9 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S32 | 45 | M | BALAGHAT | NA | 9 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S33 | 70 | F | BALAGHAT | NA | 12 April 2021 | Throat swab | Symptomatic |
| ICMR-NIRTH-S34 | 54 | M | BALAGHAT | NA | 13 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S35 | 41 | M | BALAGHAT | NA | 18 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S36 | 18 | M | BALAGHAT | NA | 21 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S37 | 45 | F | BALAGHAT | NA | 24 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S38 | 26 | F | BALAGHAT | NA | 3 February 2022 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S39 | 40 | F | BALAGHAT | NA | 4 February 2022 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S40 | 40 | M | BALAGHAT | NA | 20 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S41 | 25 | M | BALAGHAT | NA | 21 April 2021 | Throat swab | Asymptomatic |
| ICMR-NIRTH-S42 | 16 | F | BALAGHAT | NA | 27 April 2021 | Throat swab | Asymptomatic |
3. Results
3.1. Genome Assembly, Lineage Assignment, and Mutational Analysis
3.2. Effect of Novel Mutations on Protein Stability
3.3. Phylogenetic Analysis
3.4. Distribution of Circulating Lineages
4. Discussion
Limitations of This Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Ho, Y.-C. SARS-CoV-2: A Storm Is Raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef]
- Misra, G.; Manzoor, A.; Chopra, M.; Upadhyay, A.; Katiyar, A.; Bhushan, B.; Anvikar, A. Genomic Epidemiology of SARS-CoV-2 from Uttar Pradesh, India. Sci. Rep. 2023, 13, 14847. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Cases | WHO COVID-19 Dashboard. Available online: https://data.who.int/dashboards/covid19/cases?n=c (accessed on 4 June 2024).
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.A.; Areekal, B.; Rajesh, K.R.; Krishnan, J.; Suryakala, R.; Krishnan, B.; Muraly, C.P.; Santhosh, P.V. First Confirmed Case of COVID-19 Infection in India: A Case Report. Indian J. Med. Res. 2020, 151, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, A.; Gupta, A.; Joshi, M.; Aggarwal, A.; Soni, N.; Sana; Jain, D.; Verma, P.; Khandelwal, D.; et al. Demographic Comparison of the First, Second and Third Waves of COVID-19 in a Tertiary Care Hospital at Jaipur, India. Lung India 2022, 39, 525. [Google Scholar] [CrossRef] [PubMed]
- India COVID—Coronavirus Statistics—Worldometer. Available online: https://www.worldometers.info/coronavirus/country/india/ (accessed on 2 July 2024).
- Pattabiraman, C.; Habib, F.; Harsha, P.K.; Rasheed, R.; Prasad, P.; Reddy, V.; Dinesh, P.; Damodar, T.; Hosallimath, K.; George, A.K.; et al. Genomic Epidemiology Reveals Multiple Introductions and Spread of SARS-CoV-2 in the Indian State of Karnataka. PLoS ONE 2020, 15, e0243412. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, C.; Desai, A.; Prasad, P.; George, A.K.; Sreenivas, D.; Rasheed, R.; Reddy, N.V.K.; Vasanthapuram, R. Importation, Circulation, and Emergence of Variants of SARS-CoV-2 in the South Indian State of Karnataka. Wellcome Open Res 2021, 6. [Google Scholar] [CrossRef]
- Singh, B.; Avula, K.; Chatterjee, S.; Datey, A.; Ghosh, A.; De, S.; Keshry, S.S.; Ghosh, S.; Suryawanshi, A.R.; Dash, R.; et al. Isolation and Characterization of Five Severe Acute Respiratory Syndrome Coronavirus 2 Strains of Different Clades and Lineages Circulating in Eastern India. Front. Microbiol. 2022, 13, 856913. [Google Scholar] [CrossRef]
- Radhakrishnan, C.; Divakar, M.K.; Jain, A.; Viswanathan, P.; Bhoyar, R.C.; Jolly, B.; Imran, M.; Sharma, D.; Rophina, M.; Ranjan, G.; et al. Initial Insights into the Genetic Epidemiology of SARS-CoV-2 Isolates from Kerala Suggest Local Spread from Limited Introductions. Front. Genet. 2021, 12, 630542. [Google Scholar] [CrossRef]
- Joshi, M.; Puvar, A.; Kumar, D.; Ansari, A.; Pandya, M.; Raval, J.; Patel, Z.; Trivedi, P.; Gandhi, M.; Pandya, L.; et al. Genomic Variations in SARS-CoV-2 Genomes From Gujarat: Underlying Role of Variants in Disease Epidemiology. Front. Genet. 2021, 12, 586569. [Google Scholar] [CrossRef] [PubMed]
- Limaye, S.; Kasibhatla, S.M.; Ramtirthkar, M.; Kinikar, M.; Kale, M.M.; Kulkarni-Kale, U. Circulation and Evolution of SARS-CoV-2 in India: Let the Data Speak. Viruses 2021, 13, 2238. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Sabarinathan, R.; Bala, P.; Donipadi, V.; Vashisht, D.; Katika, M.R.; Kandakatla, M.; Mitra, D.; Dalal, A.; Bashyam, M.D. A Comprehensive Profile of Genomic Variations in the SARS-CoV-2 Isolates from the State of Telangana, India. J. Gen. Virol. 2021, 102, 1562. [Google Scholar] [CrossRef] [PubMed]
- MoHFW | Home. Available online: https://www.mohfw.gov.in/#latest-update (accessed on 6 June 2024).
- Andrew, S. FastQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 March 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Chen, C.W.; Lin, M.H.; Liao, C.C.; Chang, H.P.; Chu, Y.W. IStable 2.0: Predicting Protein Thermal Stability Changes by Integrating Various Characteristic Modules. Comput. Struct. Biotechnol. J. 2020, 18, 622–630. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M. Genomic Epidemiology of Novel Coronavirus: Global Subsampling. Nextstrain: Real-Time Tracking of Pathogen Evolution, 2020. [Google Scholar]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Blair Hedges, S. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Gubbay, J.; Buchan, S.A.; Daneman, N.; Mishra, S.; Patel, S.; Day, T. Inflection in Prevalence of SARS-CoV-2 Infections Missing the N501Y Mutation as a Marker of Rapid Delta (B.1.617.2) Lineage Expansion in Ontario, Canada. medRxiv 2021. [Google Scholar] [CrossRef]
- Mohapatra, R.K.; Tiwari, R.; Sarangi, A.K.; Sharma, S.K.; Khandia, R.; Saikumar, G.; Dhama, K. Twin Combination of Omicron and Delta Variants Triggering a Tsunami Wave of Ever High Surges in COVID-19 Cases: A Challenging Global Threat with a Special Focus on the Indian Subcontinent. J. Med. Virol. 2022, 94, 1761–1765. [Google Scholar] [CrossRef]
- Ray, S.K.; Mukherjee, S. Divulging Incipient SARS-CoV-2 Delta (B.1.617.2) Variant: Possible with Global Scenario. Infect. Disord. Drug Targets 2022, 23, 35–38. [Google Scholar] [CrossRef]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional Importance of the D614G Mutation in the SARS-CoV-2 Spike Protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. [Google Scholar] [CrossRef]
- Khatri, R.; Siddqui, G.; Sadhu, S.; Maithil, V.; Vishwakarma, P.; Lohiya, B.; Goswami, A.; Ahmed, S.; Awasthi, A.; Samal, S. Intrinsic D614G and P681R/H Mutations in SARS-CoV-2 VoCs Alpha, Delta, Omicron and Viruses with D614G plus Key Signature Mutations in Spike Protein Alters Fusogenicity and Infectivity. Med. Microbiol. Immunol. 2023, 212, 103–122. [Google Scholar] [CrossRef]
- Franceschi, V.B.; Caldana, G.D.; de Menezes Mayer, A.; Cybis, G.B.; Neves, C.A.M.; Ferrareze, P.A.G.; Demoliner, M.; de Almeida, P.R.; Gularte, J.S.; Hansen, A.W.; et al. Genomic Epidemiology of SARS-CoV-2 in Esteio, Rio Grande Do Sul, Brazil. BMC Genom. 2021, 22, 371. [Google Scholar] [CrossRef]
- Ghosh, A.; Walia, S.; Rattan, R.; Kanampalliwar, A.; Jha, A.; Aggarwal, S.; Fatma, S.; Das, N.; Chayani, N.; Prasad, P.; et al. Genomic Profiles of Vaccine Breakthrough SARS-CoV-2 Strains from Odisha, India. Int. J. Infect. Dis. 2022, 119, 111–113. [Google Scholar] [CrossRef]
- Ferreira, I.A.T.M.; Kemp, S.A.; Datir, R.; Saito, A.; Meng, B.; Rakshit, P.; Takaori-Kondo, A.; Kosugi, Y.; Uriu, K.; Kimura, I.; et al. SARS-CoV-2 B.1.617 Mutations L452R and E484Q Are Not Synergistic for Antibody Evasion. J Infect Dis 2021, 224, 989–994. [Google Scholar] [CrossRef]
- Pathak, A.K.; Mishra, G.P.; Uppili, B.; Walia, S.; Fatihi, S.; Abbas, T.; Banu, S.; Ghosh, A.; Kanampalliwar, A.; Jha, A.; et al. Spatio-Temporal Dynamics of Intra-Host Variability in SARS-CoV-2 Genomes. Nucleic Acids Res 2022, 50, 1551–1561. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, 2104241118. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.M.; Chakrabarti, J.; Mandal, S. Non-Synonymous Mutations of SARS-CoV-2 Leads Epitope Loss and Segregates Its Variants. Microbes Infect. 2020, 22, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J.D.; et al. The Origins and Potential Future of SARS-CoV-2 Variants of Concern in the Evolving COVID-19 Pandemic. Curr. Biol. 2021, 31, R918–R929. [Google Scholar] [CrossRef] [PubMed]
- Bano, I.; Sharif, M.; Alam, S. Genetic Drift in the Genome of SARS-CoV-2 and Its Global Health Concern. J. Med. Virol. 2022, 94, 88–98. [Google Scholar] [CrossRef] [PubMed]
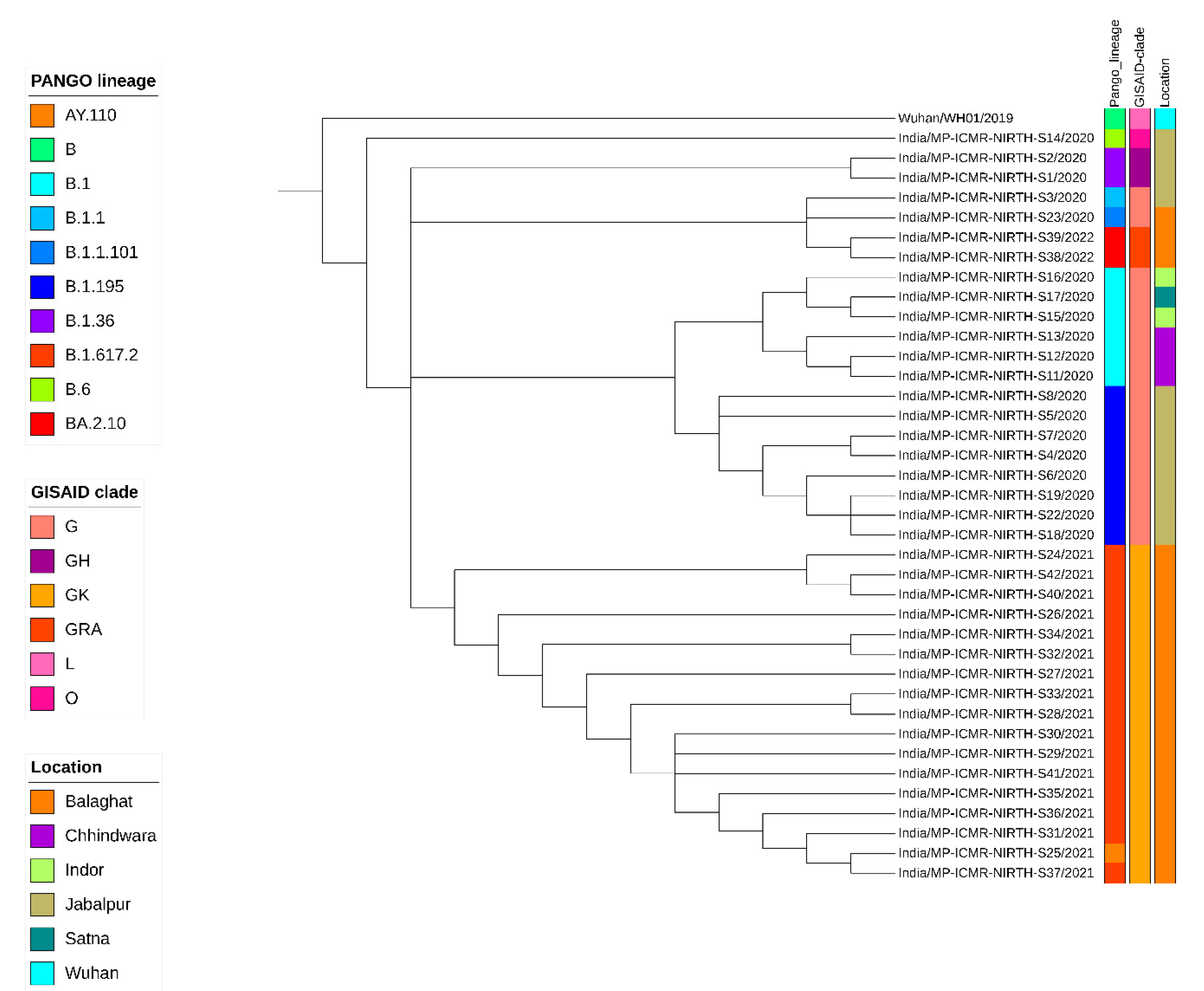
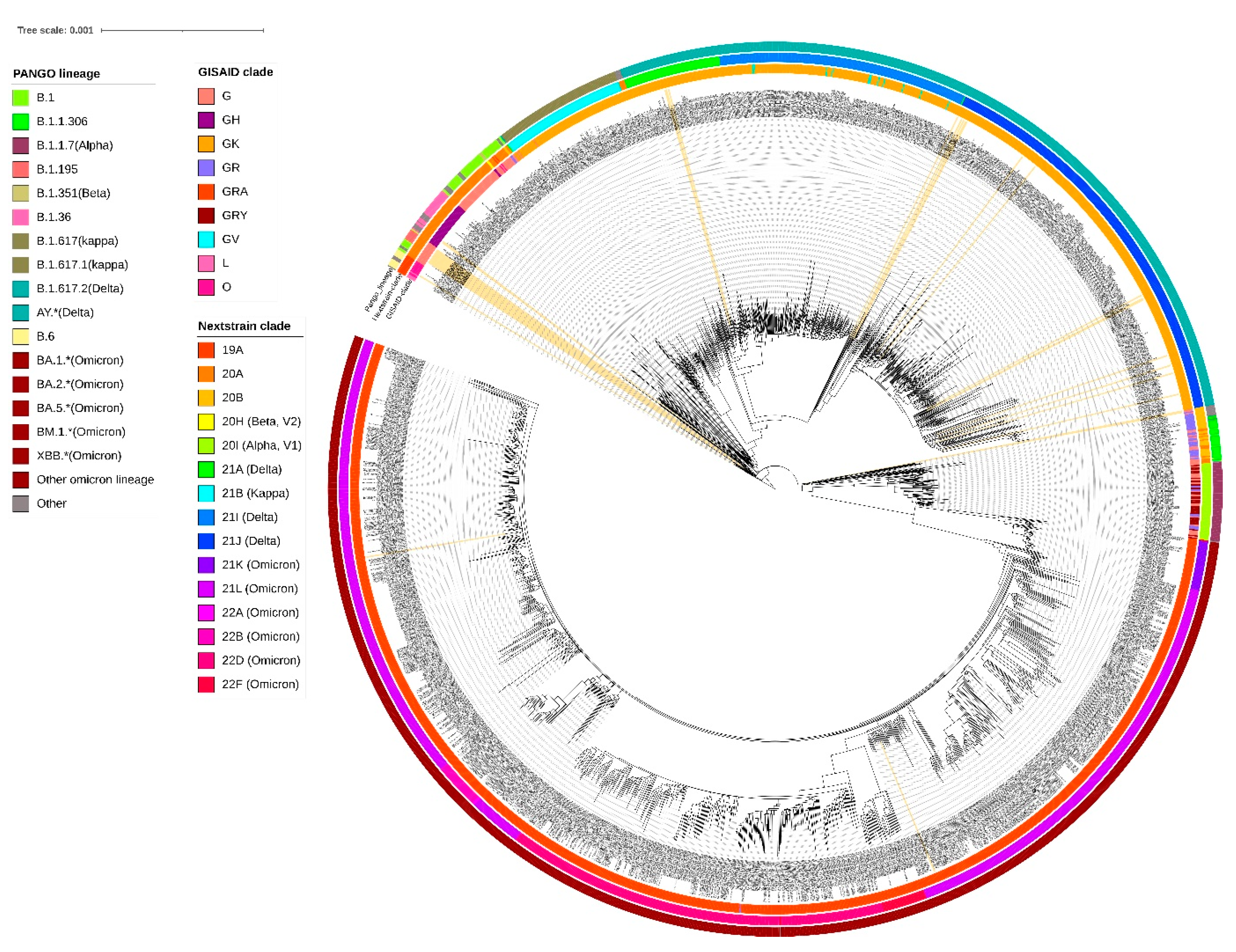
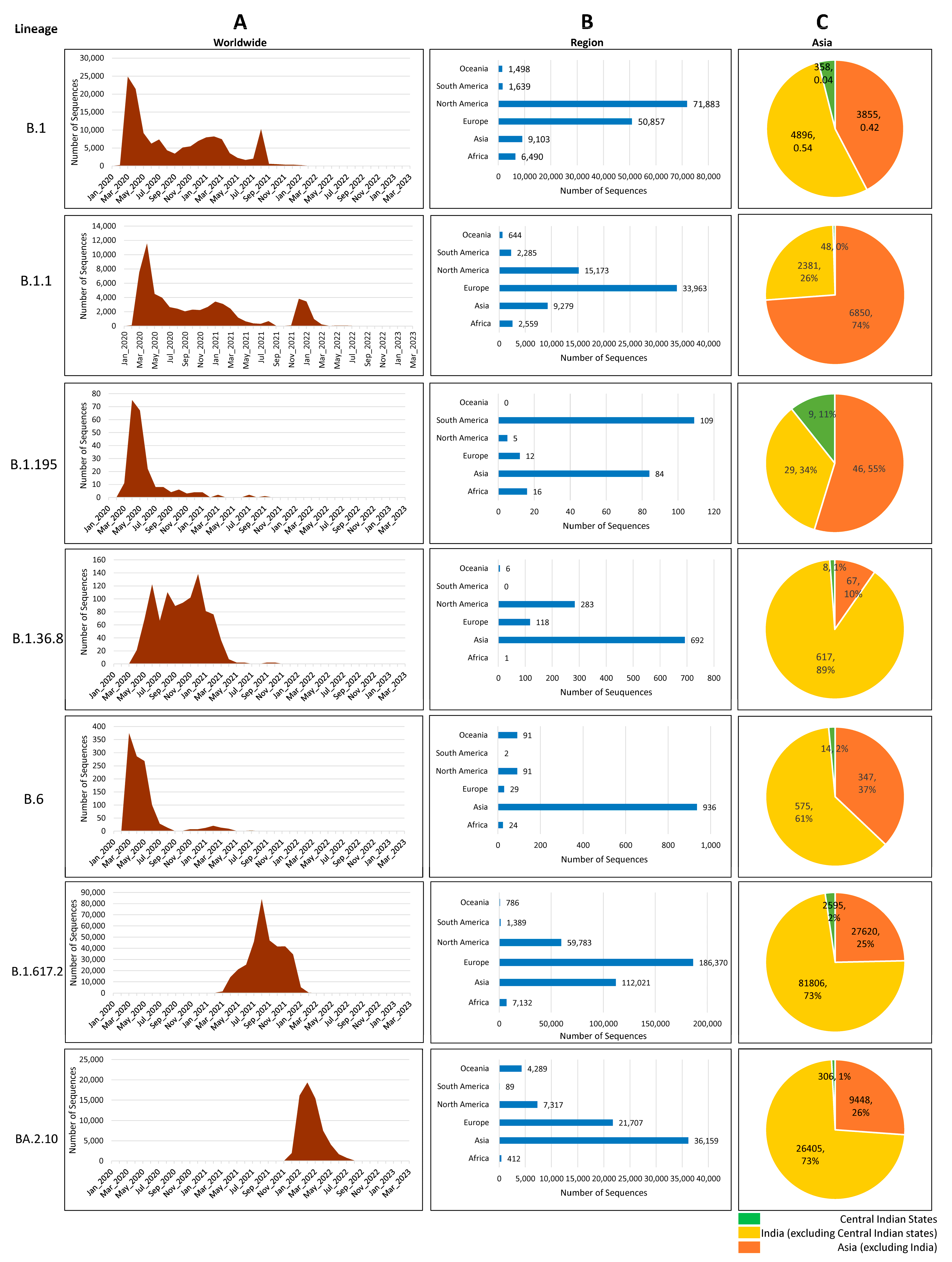
| Sr No | Sample ID | Date of Sample Collection | GISAID Clade | Nextstrain Clade | Pangolin Lineage | WHO Name |
|---|---|---|---|---|---|---|
| 1 | ICMR-NIRTH-S1 | 27 March 2020 | GH | 20A | B.1.36.8 | unassigned |
| 2 | ICMR-NIRTH-S2 | 27 March 2020 | GH | 20A | B.1.36.8 | unassigned |
| 3 | ICMR-NIRTH-S3 | 20 March 2020 | G | 20B | B.1.1 | unassigned |
| 4 | ICMR-NIRTH-S4 | 20 March 2020 | G | 20A | B.1.195 | unassigned |
| 5 | ICMR-NIRTH-S5 | 20 March 2020 | G | 20A | B.1.195 | unassigned |
| 6 | ICMR-NIRTH-S6 | 20 March 2020 | G | 20A | B.1.195 | unassigned |
| 7 | ICMR-NIRTH-S7 | 21 March 2020 | G | 20A | B.1.195 | unassigned |
| 8 | ICMR-NIRTH-S8 | 22 March 2020 | G | 20A | B.1.195 | unassigned |
| 9 | ICMR-NIRTH-S11 | 1 April 2020 | G | 20A | B.1 | unassigned |
| 10 | ICMR-NIRTH-S12 | 7 April 2020 | G | 20A | B.1 | unassigned |
| 11 | ICMR-NIRTH-S13 | 7 April 2020 | G | 20A | B.1 | unassigned |
| 12 | ICMR-NIRTH-S14 | 8 April 2020 | O | 19A | B.6 | unassigned |
| 13 | ICMR-NIRTH-S15 | 10 April 2020 | G | 20A | B.1 | unassigned |
| 14 | ICMR-NIRTH-S16 | 10 April 2020 | G | 20A | B.1 | unassigned |
| 15 | ICMR-NIRTH-S17 | 10 April 2020 | G | 20A | B.1 | unassigned |
| 16 | ICMR-NIRTH-S18 | 11 April 2020 | G | 20A | B.1.195 | unassigned |
| 17 | ICMR-NIRTH-S19 | 14 April 2020 | G | 20A | B.1.195 | unassigned |
| 18 | ICMR-NIRTH-S22 | 17 April 2020 | G | 20A | B.1.195 | unassigned |
| 19 | ICMR-NIRTH-S23 | 27 July 2020 | G | 20B | B.1.1.101 | unassigned |
| 20 | ICMR-NIRTH-S24 | 16 April 2021 | GK | 21A | B.1.617.2 | Delta |
| 21 | ICMR-NIRTH-S25 | 16 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 22 | ICMR-NIRTH-S26 | 17 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 23 | ICMR-NIRTH-S27 | 18 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 24 | ICMR-NIRTH-S28 | 19 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 25 | ICMR-NIRTH-S29 | 19 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 26 | ICMR-NIRTH-S30 | 20 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 27 | ICMR-NIRTH-S31 | 9 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 28 | ICMR-NIRTH-S32 | 9 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 29 | ICMR-NIRTH-S33 | 12 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 30 | ICMR-NIRTH-S34 | 13 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 31 | ICMR-NIRTH-S35 | 18 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 32 | ICMR-NIRTH-S36 | 21 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 33 | ICMR-NIRTH-S37 | 24 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 34 | ICMR-NIRTH-S38 | 3 February 2022 | GRA | 21L | BA.2.10 | Omicron |
| 35 | ICMR-NIRTH-S39 | 4 February 2022 | GRA | 21L | BA.2.10 | Omicron |
| 36 | ICMR-NIRTH-S40 | 20 April 2021 | GK | 21A | B.1.617.2 | Delta |
| 37 | ICMR-NIRTH-S41 | 21 April 2021 | GK | 21J | B.1.617.2 | Delta |
| 38 | ICMR-NIRTH-S42 | 27 April 2021 | GK | 21A | B.1.617.2 | Delta |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dwivedi, P.; Sharma, M.; Ansari, A.; Ghosh, A.; Bishwal, S.C.; Ray, S.K.; Katiyar, M.; Kombiah, S.; Kumar, A.; Sahare, L.; et al. Molecular Characterization and Genomic Surveillance of SARS-CoV-2 Lineages in Central India. Viruses 2024, 16, 1608. https://doi.org/10.3390/v16101608
Dwivedi P, Sharma M, Ansari A, Ghosh A, Bishwal SC, Ray SK, Katiyar M, Kombiah S, Kumar A, Sahare L, et al. Molecular Characterization and Genomic Surveillance of SARS-CoV-2 Lineages in Central India. Viruses. 2024; 16(10):1608. https://doi.org/10.3390/v16101608
Chicago/Turabian StyleDwivedi, Purna, Mukul Sharma, Afzal Ansari, Arup Ghosh, Subasa C. Bishwal, Suman Kumar Ray, Manish Katiyar, Subbiah Kombiah, Ashok Kumar, Lalit Sahare, and et al. 2024. "Molecular Characterization and Genomic Surveillance of SARS-CoV-2 Lineages in Central India" Viruses 16, no. 10: 1608. https://doi.org/10.3390/v16101608
APA StyleDwivedi, P., Sharma, M., Ansari, A., Ghosh, A., Bishwal, S. C., Ray, S. K., Katiyar, M., Kombiah, S., Kumar, A., Sahare, L., Ukey, M., Barde, P. V., Das, A., & Singh, P. (2024). Molecular Characterization and Genomic Surveillance of SARS-CoV-2 Lineages in Central India. Viruses, 16(10), 1608. https://doi.org/10.3390/v16101608