
Whole-Genome Sequencing of Two Canine Herpesvirus 1 (CaHV-1) Isolates and Clinicopathological Outcomes of Infection in French Bulldog Puppies

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case and Sampling
2.2. Histopathology
2.3. RNA In Situ Hybridization
2.4. Virus Isolation
2.5. Real Time PCR
2.6. Serum Virus Neutralization Test
2.7. Sequencing, Genome Assembly and Phylogeny
3. Results
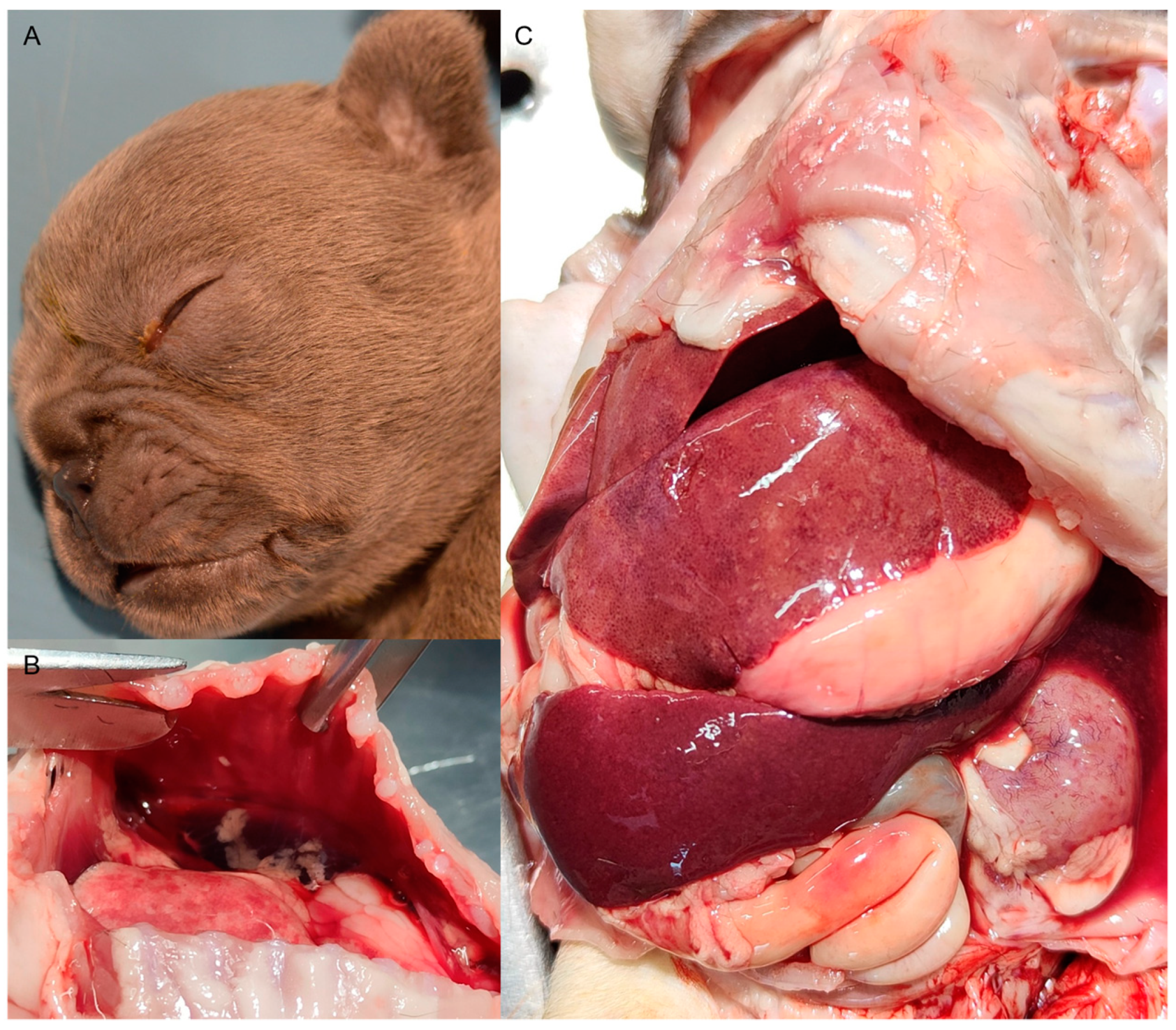
3.1. Clinical and Anatomo-Histopathological Findings, and RNA In Situ Localization
3.2. Virus Isolation
3.3. Virological Analyses
3.4. Serological Analyses
3.5. Reads Preprocessing
3.6. Genome Assembly
3.7. Phylogenetic Analysis
3.8. Variant Analysis
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lewin, A.C.; Coghill, L.M.; Mironovich, M.; Liu, C.C.; Carter, R.T.; Ledbetter, E.C. Phylogenomic analysis of global isolates of canid alphaherpesvirus 1. Viruses 2020, 12, 1421. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Castro, M.; Moraes David, M.B.; Costa Gonçalves, E.; Santos Siqueira, A.; Rodrigues Virgulino, R.; Figueira Aguiar, D.C. First molecular detection of canine herpesvirus 1 (CaHV-1) in the Eastern Brazilian Amazon. J. Vet. Sci. 2022, 23, e18. [Google Scholar] [CrossRef] [PubMed]
- Rezaeia, M.; Maziar, J.; Alizadehc, R.; Khalilib, M.; Babaeia, H. First molecular study of Canine herpesvirus-1 in reproductive specimens of adult dogs in southeast of Iran. Comp. Immunol. Microbiol. Infect. Dis. 2020, 71, 101487. [Google Scholar] [CrossRef]
- Ledbetter, E.C. Canine herpesvirus-1 ocular diseases of mature dogs. N. Z. Vet. J. 2013, 61, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Karpas, A.; Garcia, F.G.; Calvo, F.; Cross, R.E. Experimental Production of Canine Tracheobronchitis (KennelCough) with Canine Herpesvirus Isolated from Naturally Infected Dogs. Am. J. Vet. Res. 1968, 29, 1251–1257. [Google Scholar] [PubMed]
- Wright, N.G.; Cornwell, H.J. Susceptibility of 6-Week-Old Puppies to Canine Herpes Virus. J. Small. Anim. Pract. 1970, 10, 669–674. [Google Scholar] [CrossRef]
- Poste, G.; King, N. Isolation of a Herpesvirus from Canine Genital Tract—Association with Infertility, Abortion and Stillbirths. Vet. Rec. 1971, 88, 229. [Google Scholar] [CrossRef]
- Hashimoto, A.; Hirai, K.; Fukushi, H.; Fujimoto, Y. The Vaginal Lesions of a Bitch with a History of Canine Herpesvirus-Infection. Jpn. J. Vet. Sci. 1983, 45, 123–126. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Papageorgiou, K.V.; Suárez, N.M.; Wilkie, G.S.; McDonald, M.; Graham, E.M.; Davison, A.J. Genome Sequence of Canine Herpesvirus. PLoS ONE 2016, 11, e0156015. [Google Scholar] [CrossRef]
- Kurissio, J.K. Isolamento Viral e Diagnóstico Molecular de Herpevírus Canino. Master’s Thesis, Universidade Estadual Paulista, São Paulo, Brazil, 2013. [Google Scholar]
- Sarker, S.; Das, S.; Helbig, K.; Peters, A.; Raida, S.R. Genome sequence of an Australian strain of canid alphaherpesvirus 1. Aust. Vet. J. 2018, 96, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A Deubiquitinating enzyme encoded by HSV-1 belongs to a family of Cysteine Proteases that is conserved across the family Herpesviridae. Mol. Cell. 2005, 19, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.H.; Bauerfeind, R.; Sodeika, B. The C Terminus of the Large Tegument Protein pUL36 Contains Multiple Capsid Binding Sites That Function Differently during Assembly and Cell Entry of Herpes Simplex Virus. Virol. J. 2012, 86, 3682–3700. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.R.; Welschhans, R.L.; Jayaram, H.; Stow, N.D.; Preston, V.G.; Quiocho, F.A. Structural Characterization of the UL25 DNA-Packaging Protein from Herpes Simplex Virus Type 1. Virol. J. 2006, 80, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH–gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef]
- Fan, Q.; Longnecker, R.; Connollyb, S.A. A Functional Interaction between Herpes Simplex Virus 1 Glycoprotein gH/gL Domains I and II and gD Is Defined by Using Alphaherpesvirus gH and gL Chimeras. Virol. J. 2015, 89, 7159–7169. [Google Scholar] [CrossRef]
- Muylaert, I.; Zhao, Z.; Andersson, T.; Elias, P. Identification of Conserved Amino Acids in the Herpes Simplex Virus Type 1 UL8 Protein Required for DNA Synthesis and UL52 Primase Interaction in the Virus Replisome. J. Biological. Chem. 2012, 287, 33142–33152. [Google Scholar] [CrossRef]
- Kawakami, K.; Ogawa, H.; Maeda, K.; Imai, A.; Ohashi, E.; Matsunaga, S.; Tohya, Y.; Ohshima, T.; Mochizuki, M. Nosocomial Outbreak of Serious Canine Infectious Tracheobronchitis (Kennel Cough) Caused by Canine Herpesvirus Infection. J. Clin. Microbiol. 2010, 48, 1176–1181. [Google Scholar] [CrossRef]
- Decaro, N.; Amorisco, F.; Desario, C.; Lorusso, E.; Camero, M.; Bellacicco, A.L.; Sciarretta, R.; Lucente, M.S.; Martella, V.; Buonavoglia, C. Development and validation of a real-time PCR assay for specific and sensitive detection of canid herpesvirus 1. J. Virol. Methods. 2010, 169, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Rota, A.; Dogliero, A.; Biosa, T.; Messina, M.; Pregel, P.; Masoero, L. Seroprevalence of Canine Herpesvirus-1 in Breeding Dogs with or Without Vaccination in Northwest Italy. Animals 2020, 10, 1116. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 June 2022).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbinet, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Seemann, T. Prokka rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- GitHub—Tseemann/Snippy: Rapid Haploid Variant Calling and Core Genome Alignment. 2022. Available online: https://github.com/tseemann/snippy (accessed on 11 July 2022).
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyc, S. Automate assembly scaffolding elevates a new tomato system for high-throughput genome editing. bioRxiv 2021. bioRxiv: 2021.11.18.469135. [Google Scholar]
- Cingolani, P.; Cunningham, F.; McLaren, W.; Wang, K. Variant Annotations in VCF Format. 2015. Available online: https://snpeff.sourceforge.net/VCFannotationformat_v1.0.pdf (accessed on 20 September 2022).
- Labrunie, T.; Ducastelle, S.; Domenech, C.; Adere, F.; Morfina, F.; Froberta, E. UL23, UL30, and UL5 characterization of HSV1 clinical strains isolated from hematology department patients. Antivir. Res. 2019, 168, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Burrel, S.; Deback, C.; Agut, H.; Boutolleau, D. Genotypic characterization of UL23 thymidine kinase and UL30 DNA polymerase of clinical isolates of herpes simplex virus: Natural polymorphism and mutations associated with resistance to antivirals. Antimicrob. Agents Chemother. 2010, 54, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Thornton, K.E.; Chaudhuri, M.; Monahan, S.J.; Grinstead, L.A.; Parris, D.S. Analysis of in vitro activities of herpes simplex virus type 1 UL42 mutant proteins: Correlation with in vivo function. Virology 2000, 275, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Zuccola, H.J.; Filman, D.J.; Coen, D.M.; Hogle, J.M. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell. 2000, 5, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Bermek, O.; Williams, R.S. The three-component helicase/primase complex of herpes simplex virus-1. Open. Biol. 2021, 11, 210011. [Google Scholar] [CrossRef]
- Larsen, R.W.; Kiupel, M.; Balzer, H.J.; Agerholm, J.S. Prevalence of canid herpesvirus-1 infection in stillborn and dead neonatal puppies in Denmark. Acta Vet. Scand. 2015, 57, 1. [Google Scholar] [CrossRef]
- Ronsse, V.; Verstegen, J.; Onclin, K.; Farnir, F.; Poulet, H. Risk factors and reproductive disorders associated with canine herpesvirus-1 (CHV-1). Theriogenology 2004, 61, 619–636. [Google Scholar] [CrossRef]
- Reagan, K.L.; Sykes, J.E. Canine Infectious Respiratory Disease. Vet. Clin. Small Anim. Pract. 2020, 50, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Bottinelli, M.; Rampacci, E.; Stefanetti, V.; Marenzoni, M.L.; Malmlov, A.M.; Coletti, M.; Passamonti, F. Serological and biomolecular survey on canine herpesvirus-1 infection in a dog breeding kennel. J. Vet. Med. Sci. 2006, 78, 797–802. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | OP997219 | OP997220 |
|---|---|---|
| Number of Contigs | 1 | 1 |
| N50 | 125,214 | 125,214 |
| L50 | 1 | 1 |
| %GC | 31.59 | 31.59 |
| Avg. Coverage Depth | 492 | 1172 |
| Number of N | 7 | 11 |
| Predicted Genes | 77 | 77 |
| Annotated Genes | 73 | 73 |
| Location (bp) | Unit Sequence (5′–3′) | Length (bp) | Unit (bp) | Unit No. | Partial Unit (bp) |
|---|---|---|---|---|---|
| 130–227 | TCCCATAACCCC | 98 | 12 | 8 | 2 |
| 16,288–16,489 | TATCAACACCCGCGGAGAACAAGCTCCAGAATTCAGTCC ACGGAGTCTGAGTCTAATTTTGAC | 202 | 63 | 3 | 13 |
| 35,409–35,680 | AAACAACCAACCACAGTCCAGCAACCCGCC | 272 | 30 | 9 | 2 |
| 36,205–36,315 | CCAAGACCCTCAGCGTCCCAGAGT | 111 | 24 | 4 | 15 |
| 36,316–36,622 | ACGGGGAACCCGAGGACGCCAGCG | 307 | 24 | 12 | 19 |
| 62,849–62,994 | TACTGGGATCGGGGGGTTGAGGACGCGGATGGTTCACGG CACGCCGGATCGAGCGTGA | 146 | 58 | 2 | 30 |
| 93,005–93,420 | ACTATTAGAATTAACACTCTTACGTCTAGATTGTTTCAACTCTGATGCATCTCCCAACTTCTCTGTAGAATA | 407 | 72 | 5 | 47 |
| 97,625–97,773 | ATAGTCCAACCCCCTTAGGCCCCGCCCACTCAAT | 149 | 34 | 4 | 13 |
| 125,110–124,962 | |||||
| 102,512–102,679 | CATGTTGATCCTCCCTCTTTGTGTATCCCATTTGCGTGTGTAATTAGGCCGC | 168 | 52 | 3 | 12 |
| 120,223–120,056 | |||||
| 102,688–103,066 | ATATTTAAATTGGCTGCCATGTAAACCCTCCCTCTATTACGTGTGTAATTTAC | 379 | 53 | 7 | 8 |
| 120,047–119,669 | |||||
| 103,035–103,321 | CCCTCTATTACGTGTGTAATTTACATATTTAATTGAATCA | 287 | 40 | 7 | 7 |
| 119,700–119,414 | |||||
| 105,508–105,765 | AAATCTATGAATG | 257 | 13 | 19 | 10 |
| 117,203–116,970 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocchigiani, A.M.; Bertoldi, L.; Coradduzza, E.; Lostia, G.; Pintus, D.; Scivoli, R.; Cancedda, M.G.; Fiori, M.S.; Bechere, R.; Murtino, A.P.; et al. Whole-Genome Sequencing of Two Canine Herpesvirus 1 (CaHV-1) Isolates and Clinicopathological Outcomes of Infection in French Bulldog Puppies. Viruses 2024, 16, 209. https://doi.org/10.3390/v16020209
Rocchigiani AM, Bertoldi L, Coradduzza E, Lostia G, Pintus D, Scivoli R, Cancedda MG, Fiori MS, Bechere R, Murtino AP, et al. Whole-Genome Sequencing of Two Canine Herpesvirus 1 (CaHV-1) Isolates and Clinicopathological Outcomes of Infection in French Bulldog Puppies. Viruses. 2024; 16(2):209. https://doi.org/10.3390/v16020209
Chicago/Turabian StyleRocchigiani, Angela Maria, Loris Bertoldi, Elisabetta Coradduzza, Giada Lostia, Davide Pintus, Rosario Scivoli, Maria Giovanna Cancedda, Mariangela Stefania Fiori, Roberto Bechere, Anna Pina Murtino, and et al. 2024. "Whole-Genome Sequencing of Two Canine Herpesvirus 1 (CaHV-1) Isolates and Clinicopathological Outcomes of Infection in French Bulldog Puppies" Viruses 16, no. 2: 209. https://doi.org/10.3390/v16020209
APA StyleRocchigiani, A. M., Bertoldi, L., Coradduzza, E., Lostia, G., Pintus, D., Scivoli, R., Cancedda, M. G., Fiori, M. S., Bechere, R., Murtino, A. P., Pala, G., Cardeti, G., Macioccu, S., Dettori, M. A., Pintore, A., Ligios, C., & Puggioni, G. (2024). Whole-Genome Sequencing of Two Canine Herpesvirus 1 (CaHV-1) Isolates and Clinicopathological Outcomes of Infection in French Bulldog Puppies. Viruses, 16(2), 209. https://doi.org/10.3390/v16020209