
Natural Polymorphisms D60E and I62V Stabilize a Closed Conformation in HIV-1 Protease in the Absence of an Inhibitor or Substrate
Abstract
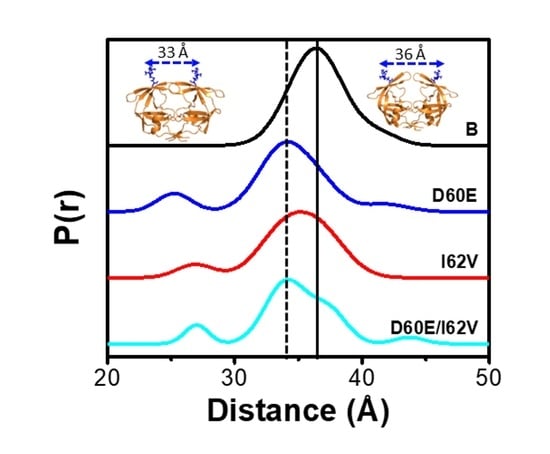
:
1. Introduction
2. Materials and Methods
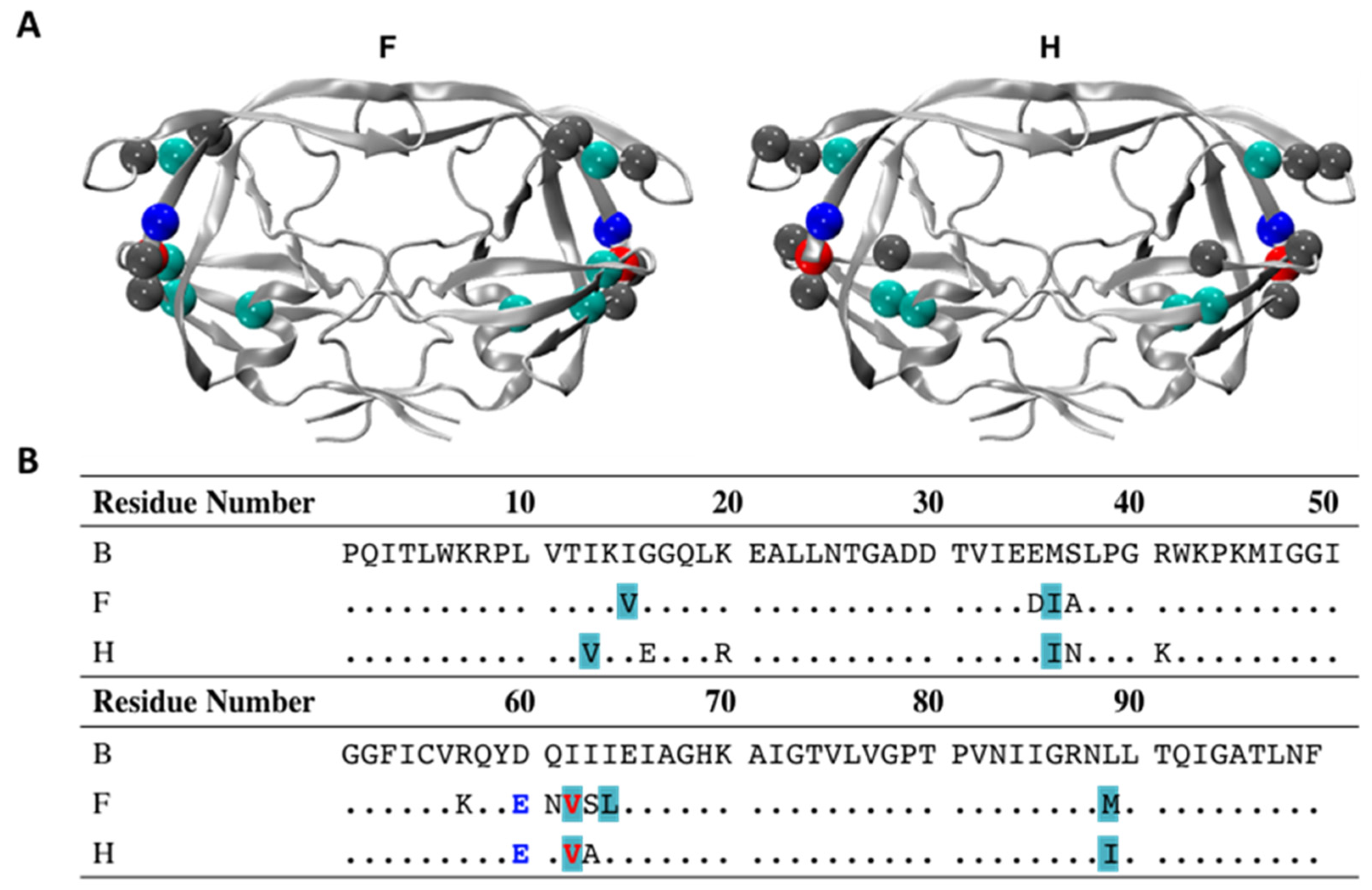
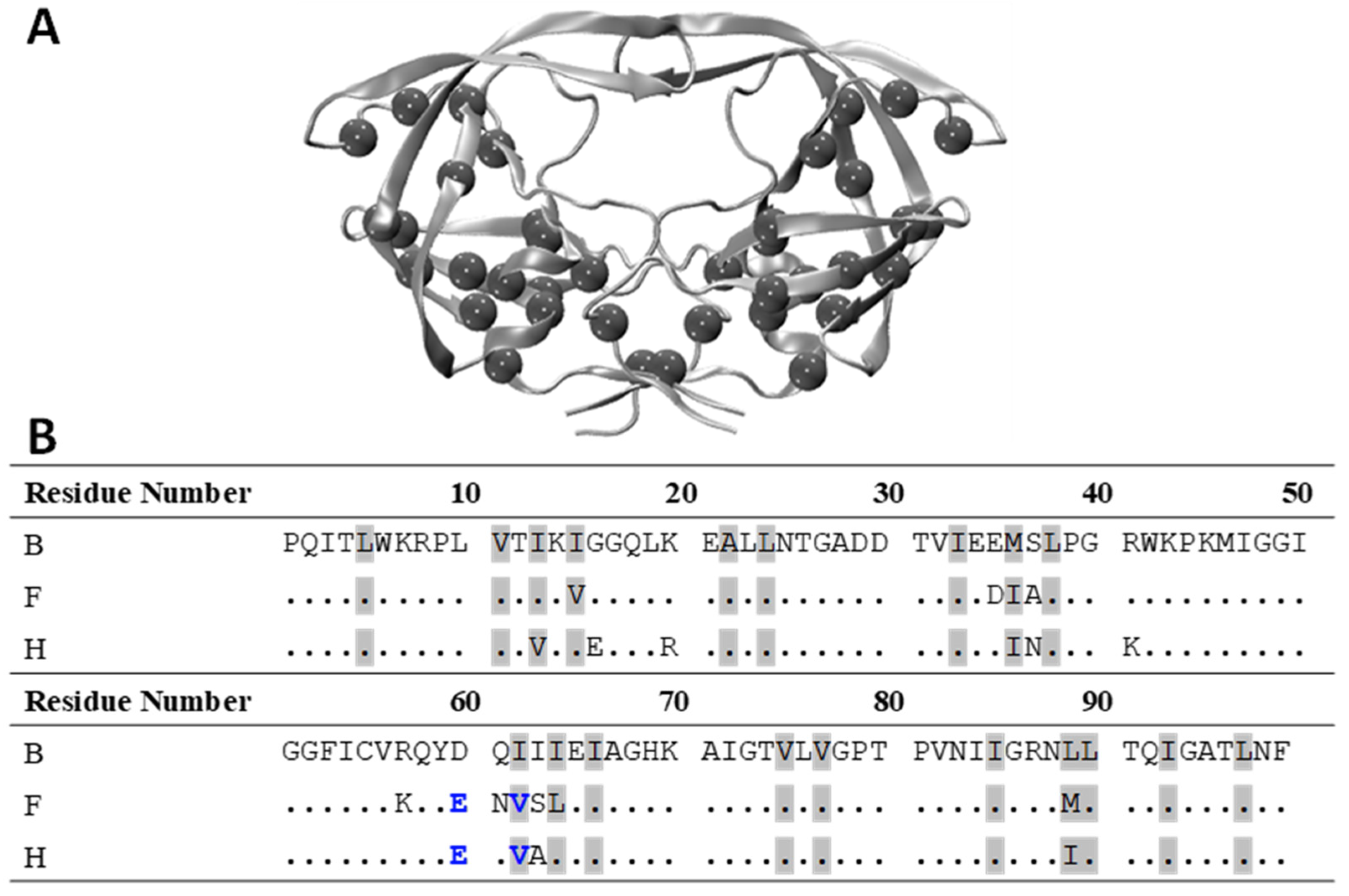
2.1. Cloning and Site-Directed Mutagenesis
2.2. Expression, Protein Purification, and Spin Labeling
2.3. DEER Experiments and Data Analysis
2.4. Molecular Dynamics Simulations
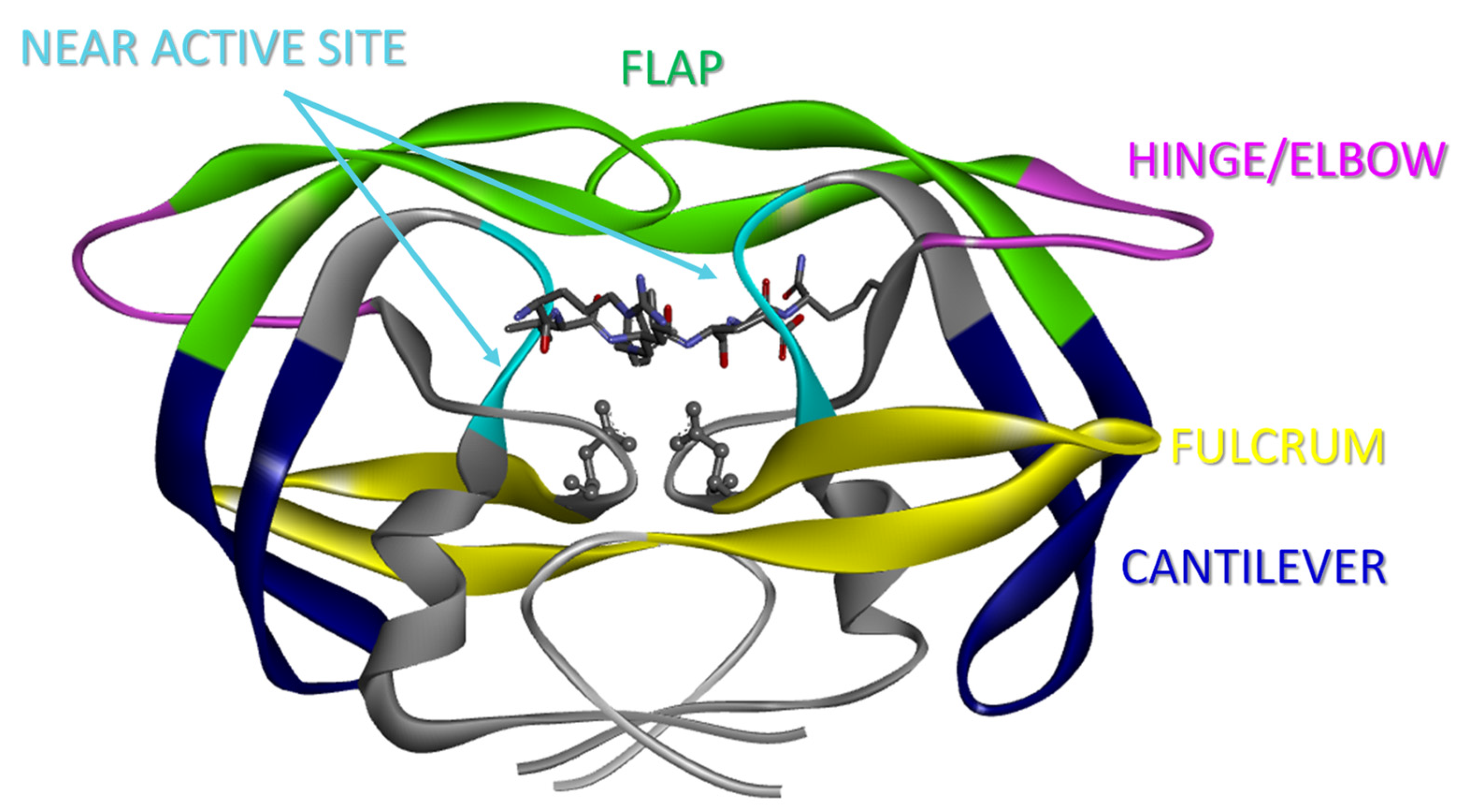
3. Results and Discussion
3.1. D60E and I62V Shift the HIV-1 PR Conformational Sampling to Closed-like States
3.2. I62V Participates in the Hydrophobic Siding Mechanism
3.3. Root-Mean-Square Deviation during Simulations
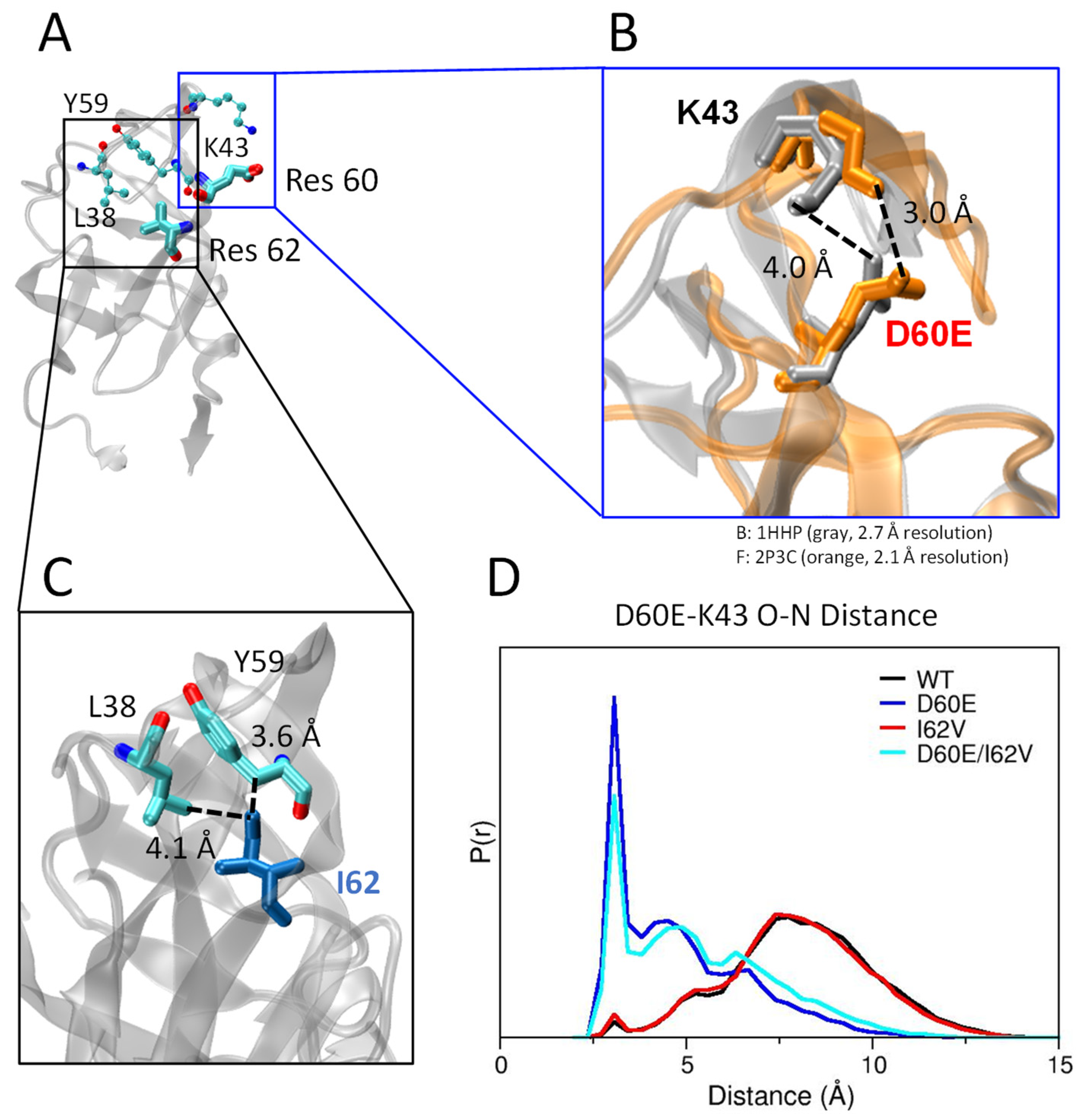
3.4. D60E Mutation Facilitates a Salt Bridge Interaction
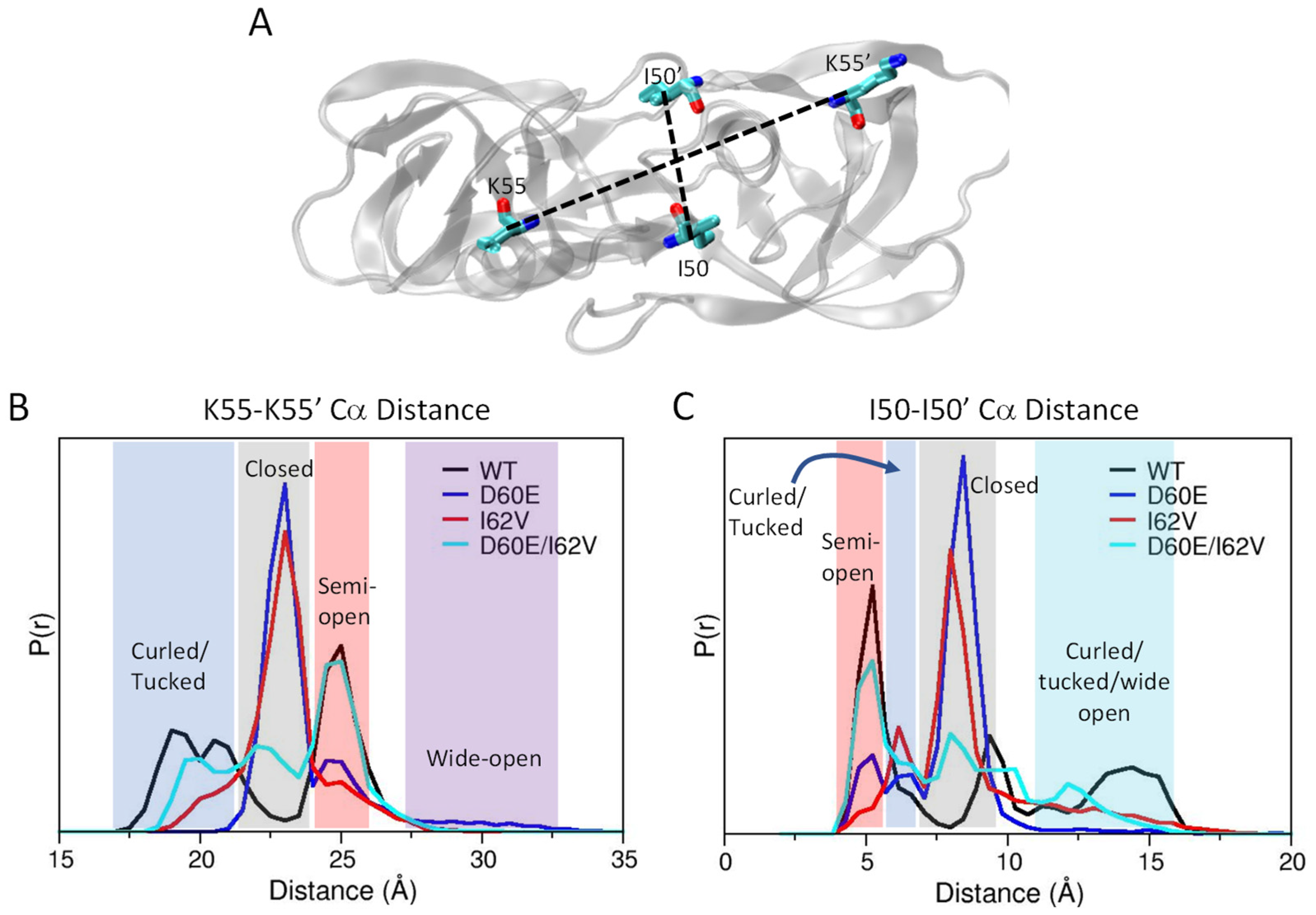
3.5. Distance Measurements for Conformational Assignment in Simulations
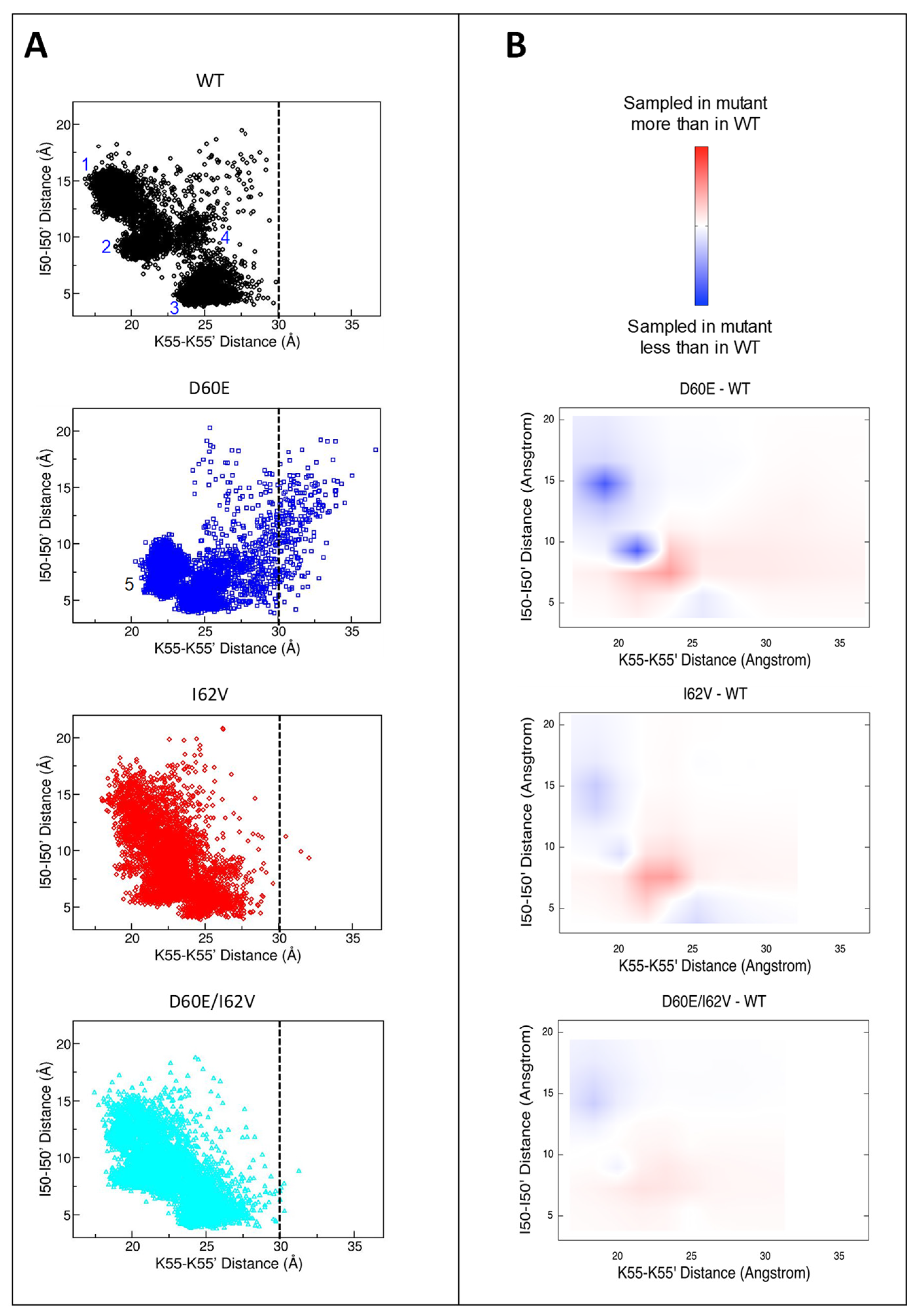
3.6. Alterations in the Conformational Landscape by Double Distance Measurement
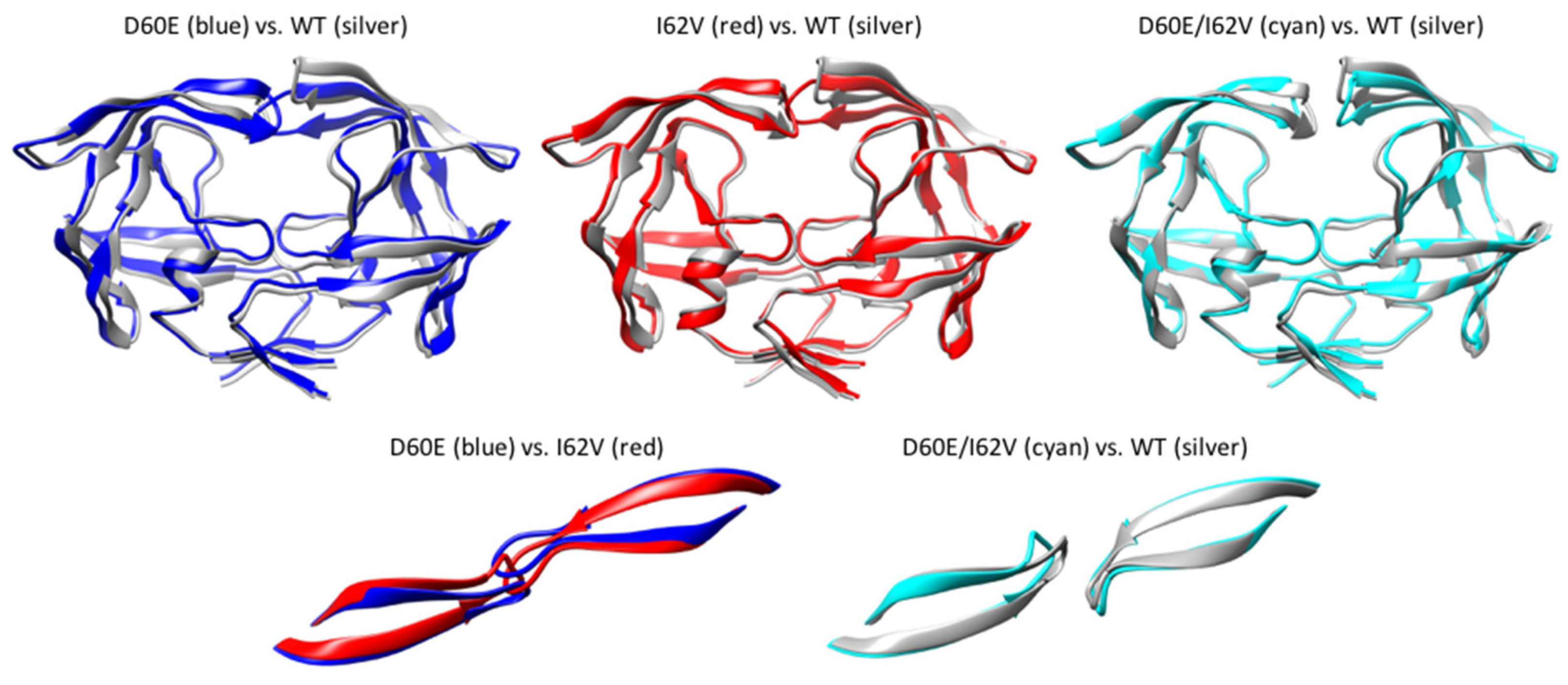
3.7. Average Structure during Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Gunthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- Walensky, R.P.; Paltiel, A.D.; Losina, E.; Mercincavage, L.M.; Schackman, B.R.; Sax, P.E.; Weinstein, M.C.; Freedberg, K.A. The survival benefits of AIDS treatment in the United States. J. Infect. Dis. 2006, 194, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cajas, J.L.; Wainberg, M.A. Protease inhibitor resistance in HIV-infected patients: Molecular and clinical perspectives. Antivir. Res. 2007, 76, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W.; Schapiro, J.M. HIV-1 drug resistance mutations: An updated framework for the second decade of HAART. Aids Rev. 2008, 10, 67–84. [Google Scholar] [PubMed]
- Weber, I.T.; Agniswamy, J. HIV-1 Protease: Structural Perspectives on Drug Resistance. Viruses 2009, 1, 1110–1136. [Google Scholar] [CrossRef] [PubMed]
- Kurt Yilmaz, N.; Swanstrom, R.; Schiffer, C.A. Improving Viral Protease Inhibitors to Counter Drug Resistance. Trends Microbiol. 2016, 24, 547–557. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef]
- Weber, I.T.; Kneller, D.W.; Wong-Sam, A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med. Chem. 2015, 7, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Gustchina, A. Structural and biochemical studies of retroviral proteases. Biochim. Biophys. Acta 2000, 1477, 16–34. [Google Scholar] [CrossRef]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef]
- UNAIDS Report on the Global AIDS Epidemic; Joint United Nations Programme on HIV/AIDS (UNAIDS): Geneva, Switzerland, 2010; pp. 19–57.
- Santos, A.F.; Soares, M.A. HIV Genetic Diversity and Drug Resistance. Viruses 2010, 2, 503–531. [Google Scholar] [CrossRef] [PubMed]
- Ashorn, P.; McQuade, T.J.; Thaisrivongs, S.; Tomasselli, A.G.; Tarpley, W.G.; Moss, B. An inhibitor of the protease blocks maturation of human and simian immunodeficiency viruses and spread of infection. Proc. Natl. Acad. Sci. USA 1990, 87, 7472–7476. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.M.; Liu, F.; Ishima, R.; Weber, I.T.; Louis, J.M. Effect of the active site D25N mutation on the structure, stability, and ligand binding of the mature HIV-1 protease. J. Biol. Chem. 2008, 283, 13459–13470. [Google Scholar] [CrossRef] [PubMed]
- Ishima, R.; Freedberg, D.I.; Wang, Y.X.; Louis, J.M.; Torchia, D.A. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure 1999, 7, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Heaslet, H.; Rosenfeld, R.; Giffin, M.; Lin, Y.C.; Tam, K.; Torbett, B.E.; Elder, J.H.; McRee, D.E.; Stout, C.D. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Liu, Q.Z.; Alzari, P.M.; Hirel, P.H.; Poljak, R.J. The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie 1991, 73, 1391–1396. [Google Scholar] [CrossRef]
- Galiano, L.; Bonora, M.; Fanucci, G.E. Inter-flap distances in HIV-1 Protease determined by pulsed EPR measurements. J. Am. Chem. Soc. 2007, 129, 11004–11005. [Google Scholar] [CrossRef]
- Freedberg, D.I.; Ishima, R.; Jacob, J.; Wang, Y.X.; Kustanovich, I.; Louis, J.M.; Torchia, D.A. Rapid structural fluctuations of the free HIV protease flaps in solution: Relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 2002, 11, 221–232. [Google Scholar] [CrossRef]
- Tozzini, V.; Trylska, J.; Chang, C.E.; McCammon, J.A. Flap opening dynamics in HIV-1 protease explored with a coarse-grained model. J. Struct. Biol. 2007, 157, 606–615. [Google Scholar] [CrossRef]
- Toth, G.; Borics, A. Flap opening mechanism of HIV-1 protease. J. Mol. Graph. Model. 2006, 24, 465–474. [Google Scholar] [CrossRef]
- Karthik, S.; Senapati, S. Dynamic flaps in HIV-1 protease adopt unique ordering at different stages in the catalytic cycle. Proteins 2011, 79, 1830–1840. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Okur, A.; Rizzo, R.C.; Simmerling, C. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2006, 103, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Layten, M.; Simmerling, C. Solution structure of HIV-1 protease flaps probed by comparison of molecular dynamics simulation ensembles and EPR experiments. J. Am. Chem. Soc. 2008, 130, 7184–7185. [Google Scholar] [CrossRef]
- de Vera, I.M.; Smith, A.N.; Dancel, M.C.; Huang, X.; Dunn, B.M.; Fanucci, G.E. Elucidating a relationship between conformational sampling and drug resistance in HIV-1 protease. Biochemistry 2013, 52, 3278–3288. [Google Scholar] [CrossRef]
- Kear, J.L.; Blackburn, M.E.; Veloro, A.M.; Dunn, B.M.; Fanucci, G.E. Subtype polymorphisms among HIV-1 protease variants confer altered flap conformations and flexibility. J. Am. Chem. Soc. 2009, 131, 14650–14651. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Casey, T.M.; Blackburn, M.E.; Huang, X.; Pham, L.; de Vera, I.M.; Carter, J.D.; Kear-Scott, J.L.; Veloro, A.M.; Galiano, L.; et al. Pulsed EPR characterization of HIV-1 protease conformational sampling and inhibitor-induced population shifts. Phys. Chem. Chem. Phys. 2016, 18, 5819–5831. [Google Scholar] [CrossRef]
- Deshmukh, L.; Louis, J.M.; Ghirlando, R.; Clore, G.M. Transient HIV-1 Gag-protease interactions revealed by paramagnetic NMR suggest origins of compensatory drug resistance mutations. Proc. Natl. Acad. Sci. USA 2016, 113, 12456–12461. [Google Scholar] [CrossRef]
- Deshmukh, L.; Tugarinov, V.; Louis, J.M.; Clore, G.M. Binding kinetics and substrate selectivity in HIV-1 protease-Gag interactions probed at atomic resolution by chemical exchange NMR. Proc. Natl. Acad. Sci. USA 2017, 114, E9855–E9862. [Google Scholar] [CrossRef]
- Agniswamy, J.; Louis, J.M.; Roche, J.; Harrison, R.W.; Weber, I.T. Structural Studies of a Rationally Selected Multi-Drug Resistant HIV-1 Protease Reveal Synergistic Effect of Distal Mutations on Flap Dynamics. PLoS ONE 2016, 11, e0168616. [Google Scholar] [CrossRef]
- Miller, M.; Schneider, J.; Sathyanarayana, B.K.; Toth, M.V.; Marshall, G.R.; Clawson, L.; Selk, L.; Kent, S.B.; Wlodawer, A. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution. Science 1989, 246, 1149–1152. [Google Scholar] [CrossRef]
- Weber, I.T.; Miller, M.; Jaskolski, M.; Leis, J.; Skalka, A.M.; Wlodawer, A. Molecular modeling of the HIV-1 protease and its substrate binding site. Science 1989, 243, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Coman, R.M.; Thiaville, M.M.; Janka, L.K.; Jeung, J.A.; Nukoolkarn, S.; Govindasamy, L.; Agbandje-McKenna, M.; McKenna, R.; Leelamanit, W.; et al. Analysis of HIV-1 CRF_01 A/E protease inhibitor resistance: Structural determinants for maintaining sensitivity and developing resistance to atazanavir. Biochemistry 2006, 45, 5468–5477. [Google Scholar] [CrossRef]
- Martin, P.; Vickrey, J.F.; Proteasa, G.; Jimenez, Y.L.; Wawrzak, Z.; Winters, M.A.; Merigan, T.C.; Kovari, L.C. “Wide-open” 1.3 A structure of a multidrug-resistant HIV-1 protease as a drug target. Structure 2005, 13, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Coman, R.M.; Robbins, A.H.; Goodenow, M.M.; Dunn, B.M.; McKenna, R. High-resolution structure of unbound human immunodeficiency virus 1 subtype C protease: Implications of flap dynamics and drug resistance. Acta Crystallogr. D Biol. Crystallogr. 2008, D64, 754–763. [Google Scholar] [CrossRef]
- Shen, C.H.; Chang, Y.C.; Agniswamy, J.; Harrison, R.W.; Weber, I.T. Conformational variation of an extreme drug resistant mutant of HIV protease. J. Mol. Graph. Model. 2015, 62, 87–96. [Google Scholar] [CrossRef]
- Wong-Sam, A.; Wang, Y.F.; Kneller, D.W.; Kovalevsky, A.Y.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. HIV-1 protease with 10 lopinavir and darunavir resistance mutations exhibits altered inhibition, structural rearrangements and extreme dynamics. J. Mol. Graph. Model. 2022, 117, 108315. [Google Scholar] [CrossRef] [PubMed]
- Souffrant, M.; Yao, X.Q.; Hamelberg, D. Evolving Mutational Buildup in HIV-1 Protease Shifts Conformational Dynamics to Gain Drug Resistance. J. Chem. Inf. Model. 2023, 63, 3892–3902. [Google Scholar] [CrossRef]
- Galiano, L.; Ding, F.; Veloro, A.M.; Blackburn, M.E.; Simmerling, C.; Fanucci, G.E. Drug pressure selected mutations in HIV-1 protease alter flap conformations. J. Am. Chem. Soc. 2009, 131, 430–431. [Google Scholar] [CrossRef]
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.; Wensing, A.M.; Richman, D.D. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2011, 19, 156–164. [Google Scholar]
- Wensing, A.M.; Calvez, V.; Gunthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2014 Update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2014, 22, 642–650. [Google Scholar]
- Muzammil, S.; Armstrong, A.A.; Kang, L.W.; Jakalian, A.; Bonneau, P.R.; Schmelmer, V.; Amzel, L.M.; Freire, E. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J. Virol. 2007, 81, 5144–5154. [Google Scholar] [CrossRef]
- Doyon, L.; Tremblay, S.; Bourgon, L.; Wardrop, E.; Cordingley, M.G. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antivir. Res. 2005, 68, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sturmer, M.; Stephan, C.; Gute, P.; Knecht, G.; Bickel, M.; Brodt, H.R.; Doerr, H.W.; Gurtler, L.; Lecocq, P.; van Houtte, M. Comparison of drug resistance scores for tipranavir in protease inhibitor-naive patients infected with HIV-1 B and non-B subtypes. Antimicrob. Agents Chemother. 2011, 55, 5362–5366. [Google Scholar] [CrossRef] [PubMed]
- Nalam, M.N.; Schiffer, C.A. New approaches to HIV protease inhibitor drug design II: Testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr. Opin. HIV AIDS 2008, 3, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Brunzelle, J.S.; Kovari, I.A.; Dewdney, T.G.; Reiter, S.J.; Kovari, L.C. The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochem. Biophys. Res. Commun. 2011, 412, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: Pathogenetic and therapeutic implications. J. Virol. 2007, 81, 10209–10219. [Google Scholar] [CrossRef] [PubMed]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Krauchenco, S.; Martins, N.H.; Sanches, M.; Polikarpov, I. Effectiveness of commercial inhibitors against subtype F HIV-1 protease. J. Enzym. Inhib. Med. Chem. 2009, 24, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Mata-Munguia, C.; Escoto-Delgadillo, M.; Torres-Mendoza, B.; Flores-Soto, M.; Vazquez-Torres, M.; Galvez-Gastelum, F.; Viniegra-Osorio, A.; Castillero-Manzano, M.; Vazquez-Valls, E. Natural polymorphisms and unusual mutations in HIV-1 protease with potential antiretroviral resistance: A bioinformatic analysis. BMC Bioinform. 2014, 15, 72. [Google Scholar] [CrossRef]
- Holguin, A.; Sune, C.; Hamy, F.; Soriano, V.; Klimkait, T. Natural polymorphisms in the protease gene modulate the replicative capacity of non-B HIV-1 variants in the absence of drug pressure. J. Clin. Virol. 2006, 36, 264–271. [Google Scholar] [CrossRef]
- Holguin, A.; Paxinos, E.; Hertogs, K.; Womac, C.; Soriano, V. Impact of frequent natural polymorphisms at the protease gene on the in vitro susceptibility to protease inhibitors in HIV-1 non-B subtypes. J. Clin. Virol. 2004, 31, 215–220. [Google Scholar] [CrossRef]
- Agniswamy, J.; Sayer, J.M.; Weber, I.T.; Louis, J.M. Terminal interface conformations modulate dimer stability prior to amino terminal autoprocessing of HIV-1 protease. Biochemistry 2012, 51, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.; Wang, C. A Single Amino Acid Substitution at the HIV-1 Protease Termini Dimer Interface Significantly Reduces Viral Particles Processing Efficiency. Jpn. J. Infect. Dis. 2021, 74, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Dakshinamoorthy, A.; Asmita, A.; Senapati, S. Comprehending the Structure, Dynamics, and Mechanism of Action of Drug-Resistant HIV Protease. ACS Omega 2023, 8, 9748–9763. [Google Scholar] [CrossRef]
- Kim, J.G.; Shan, L. Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs. Viruses 2022, 14, 1179. [Google Scholar] [CrossRef]
- Okafor, S.N.; Angsantikul, P.; Ahmed, H. Discovery of Novel HIV Protease Inhibitors Using Modern Computational Techniques. Int. J. Mol. Sci. 2022, 23, 12149. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, M.; Taghdir, M.; Abasi Joozdani, F. Dynamozones are the most obvious sign of the evolution of conformational dynamics in HIV-1 protease. Sci. Rep. 2023, 13, 14179. [Google Scholar] [CrossRef]
- Rajendran, M.; Ferran, M.C.; Mouli, L.; Babbitt, G.A.; Lynch, M.L. Evolution of drug resistance drives destabilization of flap region dynamics in HIV-1 protease. Biophys. Rep. 2023, 3, 100121. [Google Scholar] [CrossRef]
- Sheik Ismail, Z.; Worth, R.; Mosebi, S.; Sayed, Y. HIV Protease Hinge Region Insertions at Codon 38 Affect Enzyme Kinetics, Conformational Stability and Dynamics. Protein J. 2023, 42, 490–501. [Google Scholar] [CrossRef]
- Spielvogel, E.; Lee, S.K.; Zhou, S.; Lockbaum, G.J.; Henes, M.; Sondgeroth, A.; Kosovrasti, K.; Nalivaika, E.A.; Ali, A.; Yilmaz, N.K.; et al. Selection of HIV-1 for resistance to fifth-generation protease inhibitors reveals two independent pathways to high-level resistance. eLife 2023, 12, e80328. [Google Scholar] [CrossRef]
- Tunc, H.; Dogan, B.; Darendeli Kiraz, B.N.; Sari, M.; Durdagi, S.; Kotil, S. Prediction of HIV-1 protease resistance using genotypic, phenotypic, and molecular information with artificial neural networks. PeerJ 2023, 11, e14987. [Google Scholar] [CrossRef]
- Wang, Q.; Gao, H.; Clark, K.M.; Mugisha, C.S.; Davis, K.; Tang, J.P.; Harlan, G.H.; DeSelm, C.J.; Presti, R.M.; Kutluay, S.B.; et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science 2021, 371, eabe1707. [Google Scholar] [CrossRef]
- Yu, F.H.; Huang, K.J.; Wang, C.T. Conditional activation of an HIV-1 protease attenuated mutant by a leucine zipper dimerization motif. Virus Res. 2021, 295, 198258. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Shan, Q.; Ma, L.; Dong, B.; Wang, J.; Zhang, G.; Wang, M.; Zhou, J.; Cen, S.; Wang, Y. Structure based design and evaluation of benzoheterocycle derivatives as potential dual HIV-1 protease and reverse transcriptase inhibitors. Eur. J. Med. Chem. 2023, 246, 114981. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Ali, A.; Rusere, L.; Soumana, D.I.; Kurt Yilmaz, N.; Schiffer, C.A. Dengue Virus NS2B/NS3 Protease Inhibitors Exploiting the Prime Side. J. Virol. 2017, 91, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Nalivaika, E.A.; Prachanronarong, K.L.; Yilmaz, N.K.; Schiffer, C.A. Dengue Protease Substrate Recognition: Binding of the Prime Side. ACS Infect. Dis. 2016, 2, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Zephyr, J.; Kurt Yilmaz, N.; Schiffer, C.A. Viral proteases: Structure, mechanism and inhibition. Enzymes 2021, 50, 301–333. [Google Scholar] [CrossRef] [PubMed]
- Borbat, P.P.; Freed, J.H. Pulse Dipolar Electron Spin Resonance: Distance Measurements. In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences; Timmel, C.R., Harmer, J.R., Eds.; Springer: Berlin, Germany, 2014; Volume 152, pp. 1–82. [Google Scholar]
- Ghimire, H.; McCarrick, R.M.; Budil, D.E.; Lorigan, G.A. Significantly improved sensitivity of Q-band PELDOR/DEER experiments relative to X-band is observed in measuring the intercoil distance of a leucine zipper motif peptide (GCN4-LZ). Biochemistry 2009, 48, 5782–5784. [Google Scholar] [CrossRef]
- Goldfarb, D. Exploring protein conformations in vitro and in cell with EPR distance measurements. Curr. Opin. Struct. Biol. 2022, 75, 102398. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef]
- Polyhach, Y.; Bordingnon, E.; Tschaggler, S.; Gandra, A.; Godt, A.; Jeschke, G. High Sensitivity and versatality of the DEER experiment on nitroxide radical pairs at Q-band frequenies. Phys. Chem. Chem. Phys. 2012, 14, 10762–10773. [Google Scholar] [CrossRef] [PubMed]
- Reginsson, G.W.; Schiemann, O. Pulsed electron-electron double resonance: Beyond nanometre distance measurements on biomacromolecules. Biochem. J. 2011, 434, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Torricella, F.; Pierro, A.; Mileo, E.; Belle, V.; Bonucci, A. Nitroxide spin labels and EPR spectroscopy: A powerful association for protein dynamics studies. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140653. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.E.; Veloro, A.M.; Fanucci, G.E. Monitoring inhibitor-induced conformational population shifts in HIV-1 protease by pulsed EPR spectroscopy. Biochemistry 2009, 48, 8765–8767. [Google Scholar] [CrossRef] [PubMed]
- de Vera, I.M.; Blackburn, M.E.; Fanucci, G.E. Correlating conformational shift induction with altered inhibitor potency in a multidrug resistant HIV-1 protease variant. Biochemistry 2012, 51, 7813–7815. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; de Vera, I.M.; Veloro, A.M.; Blackburn, M.E.; Kear, J.L.; Carter, J.D.; Rocca, J.R.; Simmerling, C.; Dunn, B.M.; Fanucci, G.E. Inhibitor-induced conformational shifts and ligand-exchange dynamics for HIV-1 protease measured by pulsed EPR and NMR spectroscopy. J. Phys. Chem. B 2012, 116, 14235–14244. [Google Scholar] [CrossRef]
- Liu, Z.; Tran, T.T.; Pham, L.; Hu, L.; Bentz, K.; Savin, D.A.; Fanucci, G.E. Darunavir-Resistant HIV-1 Protease Constructs Uphold a Conformational Selection Hypothesis for Drug Resistance. Viruses 2020, 12, 1275. [Google Scholar] [CrossRef]
- Tran, T.T.; Liu, Z.; Fanucci, G.E. Conformational landscape of non-B variants of HIV-1 protease: A pulsed EPR study. Biochem. Biophys. Res. Commun. 2020, 532, 219–224. [Google Scholar] [CrossRef]
- Huang, X.; Britto, M.D.; Kear-Scott, J.L.; Boone, C.D.; Rocca, J.R.; Simmerling, C.; McKenna, R.; Bieri, M.; Gooley, P.R.; Dunn, B.M.; et al. The role of select subtype polymorphisms on HIV-1 protease conformational sampling and dynamics. J. Biol. Chem. 2014, 289, 17203–17214. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, X.; Hu, L.; Pham, L.; Poole, K.M.; Tang, Y.; Mahon, B.P.; Tang, W.; Li, K.; Goldfarb, N.E.; et al. Effects of Hinge-region Natural Polymorphisms on Human Immunodeficiency Virus-Type 1 Protease Structure, Dynamics, and Drug Pressure Evolution. J. Biol. Chem. 2016, 291, 22741–22756. [Google Scholar] [CrossRef]
- Carter, J.D.; Gonzales, E.G.; Huang, X.; Smith, A.N.; de Vera, I.M.; D’Amore, P.W.; Rocca, J.R.; Goodenow, M.M.; Dunn, B.M.; Fanucci, G.E. Effects of PRE and POST therapy drug-pressure selected mutations on HIV-1 protease conformational sampling. FEBS Lett. 2014, 588, 3123–3128. [Google Scholar] [CrossRef]
- Mildner, A.M.; Rothrock, D.J.; Leone, J.W.; Bannow, C.A.; Lull, J.M.; Reardon, I.M.; Sarcich, J.L.; Howe, W.J.; Tomich, C.S.; Smith, C.W. The HIV-1 protease as enzyme and substrate: Mutagenesis of autolysis sites and generation of a stable mutant with retained kinetic properties. Biochemistry 1994, 33, 9405–9413. [Google Scholar] [CrossRef]
- Kear, J.L.; Galiano, L.; Veloro, A.M.; Harris, J.; Busenlehner, L.S.; Fanucci, G.E. Monitoring the autoproteolysis of HIV-1 Protease by Site-Directed Spin-Labeling and Electron Paramagnetic Resonance Spectroscopy. J. Biophys. Chem. 2011, 2, 137–146. [Google Scholar] [CrossRef]
- Louis, J.M.; Clore, G.M.; Gronenborn, A.M. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999, 6, 868–875. [Google Scholar] [CrossRef]
- Shao, W.; Everitt, L.; Manchester, M.; Loeb, D.D.; Hutchison, C.A., 3rd; Swanstrom, R. Sequence requirements of the HIV-1 protease flap region determined by saturation mutagenesis and kinetic analysis of flap mutants. Proc. Natl. Acad. Sci. USA 1997, 94, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis2006—A Comprehensive Software Package for Analyzing Pulsed ELDOR Data. Appl. Mag. Reson. 2006, 30, 473–498. [Google Scholar] [CrossRef]
- Casey, T.M.; Fanucci, G.E. Spin labeling and Double Electron-Electron Resonance (DEER) to Deconstruct Conformational Ensembles of HIV Protease. Methods Enzym. 2015, 564, 153–187. [Google Scholar] [CrossRef]
- de Vera, I.M.S.; Blackburn, M.E.; Galiano, L.; Fanucci, G.E. Pulsed EPR distance measurements in soluble proteins by site-directed spin labeling (SDSL). Curren Protoc. Protein Sci. 2013, 74, 29. [Google Scholar] [CrossRef]
- Chakravorty, D.K.; Hammes-Schiffer, S. Impact of mutation on proton transfer reactions in ketosteroid isomerase: Insights from molecular dynamics simulations. J. Am. Chem. Soc. 2010, 132, 7549–7555. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, D.K.; Wang, B.; Ucisik, M.N.; Merz, K.M., Jr. Insight into the cation-pi interaction at the metal binding site of the copper metallochaperone CusF. J. Am. Chem. Soc. 2011, 133, 19330–19333. [Google Scholar] [CrossRef]
- Chakravorty, D.K.; Wang, B.; Lee, C.W.; Giedroc, D.P.; Merz, K.M., Jr. Simulations of allosteric motions in the zinc sensor CzrA. J. Am. Chem. Soc. 2012, 134, 3367–3376. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, D.K.; Parker, T.M.; Guerra, A.J.; Sherrill, C.D.; Giedroc, D.P.; Merz, K.M., Jr. Energetics of zinc-mediated interactions in the allosteric pathways of metal sensor proteins. J. Am. Chem. Soc. 2013, 135, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.D.; Mathias, J.D.; Gomez, E.F.; Ran, Y.; Xu, F.; Galiano, L.; Tran, N.Q.; D’Amore, P.W.; Wright, C.S.; Chakravorty, D.K.; et al. Characterizing solution surface loop conformational flexibility of the GM2 activator protein. J. Phys. Chem. B 2014, 118, 10607–10617. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.S.; Cheatham, T.E., 3rd. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Adelman, S.A.; Doll, J.D. Generalized Langevin Equation Approach for Atom/Solid-Surface Scattering: General Formulation for Classical Scattering off Harmonic Solids. J. Chem. Phys. 1976, 64, 2375–2388. [Google Scholar] [CrossRef]
- Doll, J.D.; Dion, D.R. Generalized Langevin Equation Approach for Atom/Solid Surface Scattering: Numerical Techniques for Gaussian Generalized Langevin Dynamics. J. Chem. Phys. 1976, 65, 3762–3766. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulations of Liquids; Claraden Press: Oxford, UK, 1987. [Google Scholar]
- Clemente, J.C.; Hemrajani, R.; Blum, L.E.; Goodenow, M.M.; Dunn, B.M. Secondary mutations M36I and A71V in the human immunodeficiency virus type 1 protease can provide an advantage for the emergence of the primary mutation D30N. Biochemistry 2003, 42, 15029–15035. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Moose, R.E.; Hemrajani, R.; Whitford, L.R.; Govindasamy, L.; Reutzel, R.; McKenna, R.; Agbandje-McKenna, M.; Goodenow, M.M.; Dunn, B.M. Comparing the accumulation of active- and nonactive-site mutations in the HIV-1 protease. Biochemistry 2004, 43, 12141–12151. [Google Scholar] [CrossRef] [PubMed]
- Foulkes-Murzycki, J.E.; Scott, W.R.; Schiffer, C.A. Hydrophobic sliding: A possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 2007, 15, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, N.E.; Ohanessian, M.; Biswas, S.; McGee, T.D., Jr.; Mahon, B.P.; Ostrov, D.A.; Garcia, J.; Tang, Y.; McKenna, R.; Roitberg, A.; et al. Defective hydrophobic sliding mechanism and active site expansion in HIV-1 protease drug resistant variant Gly48Thr/Leu89Met: Mechanisms for the loss of saquinavir binding potency. Biochemistry 2014, 54, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Cai, Y.; Nalam, M.N.; Bolon, D.N.; Schiffer, C.A. Hydrophobic core flexibility modulates enzyme activity in HIV-1 protease. J. Am. Chem. Soc. 2012, 134, 4163–4168. [Google Scholar] [CrossRef]
- Pillai, B.; Kannan, K.K.; Hosur, M.V. 1.9 A X-ray study shows closed flap conformation in crystals of tethered HIV-1 PR. Proteins 2001, 43, 57–64. [Google Scholar] [CrossRef]
- Robbins, A.H.; Coman, R.M.; Bracho-Sanchez, E.; Fernandez, M.A.; Gilliland, C.T.; Li, M.; Agbandje-McKenna, M.; Wlodawer, A.; Dunn, B.M.; McKenna, R. Structure of the unbound form of HIV-1 subtype A protease: Comparison with unbound forms of proteases from other HIV subtypes. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 233–242. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Most Probable Distance (±0.2 Å) | Conformation Population Percentages | ||||
|---|---|---|---|---|---|
| Curled/Tucked | Closed | Semi-Open | Open | ||
| B a | 36.2 | 4 ± 3 | 3 ± 3 | 86 ± 3 | 7 ± 3 |
| F b | 34.4 | 20 ± 4 | 49 ± 5 | 28 ± 5 | 3 ± 3 |
| H b | 33.7 | 11 ± 5 | 48 ± 5 | 41 ± 5 | 0 ± 5 |
| D60E | 34.0 | 17 ± 5 | 65 ± 5 | 12 ± 5 | 7 ± 5 |
| I62V | 35.2 | 10 ± 3 | 49 ± 3 | 41 ± 3 | 0 ± 3 |
| D60E/I63E | 34.2 and 36.5 | 10 ± 4 | 59 ± 4 | 26 ± 4 | 5 ± 4 |
| A71V c | 33.0 | 17 ± 6 | 67 ± 6 | 7 ± 6 | 9 ± 6 |
| L/I63P d | 33.2 | 10 ± 5 | 57 ± 5 | 27 ± 5 | 6 ± 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.T.; Fanucci, G.E. Natural Polymorphisms D60E and I62V Stabilize a Closed Conformation in HIV-1 Protease in the Absence of an Inhibitor or Substrate. Viruses 2024, 16, 236. https://doi.org/10.3390/v16020236
Tran TT, Fanucci GE. Natural Polymorphisms D60E and I62V Stabilize a Closed Conformation in HIV-1 Protease in the Absence of an Inhibitor or Substrate. Viruses. 2024; 16(2):236. https://doi.org/10.3390/v16020236
Chicago/Turabian StyleTran, Trang T., and Gail E. Fanucci. 2024. "Natural Polymorphisms D60E and I62V Stabilize a Closed Conformation in HIV-1 Protease in the Absence of an Inhibitor or Substrate" Viruses 16, no. 2: 236. https://doi.org/10.3390/v16020236