
Porcine Sapovirus Protease Controls the Innate Immune Response and Targets TBK1
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus
2.2. Expression Vectors
2.3. Transfection of Cell Lines
2.4. Reporter Gene Assays
2.5. 5BR Assay
2.6. Poly(I:C) Stimulation and RT-qPCR
2.7. CRISPR/Cas9-Mediated Genome Editing
2.8. Immunoprecipitation (IP)
2.9. SDS-PAGE and Immunoblotting
2.10. PSaV Infection
2.11. TCID50 Assay
2.12. Statistical Analysis and Software
3. Results
3.1. PSaV NS6 Is a Type I IFN Antagonist
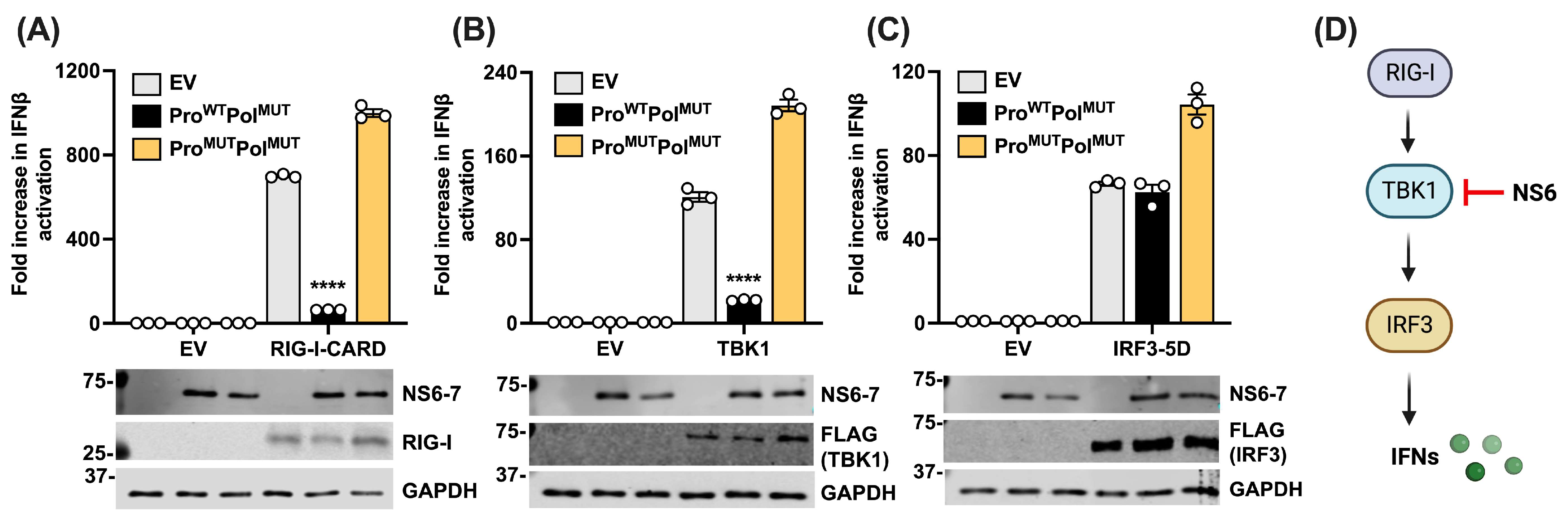
3.2. NS6 Restricts the Type I IFN Pathway at the Level of TBK1
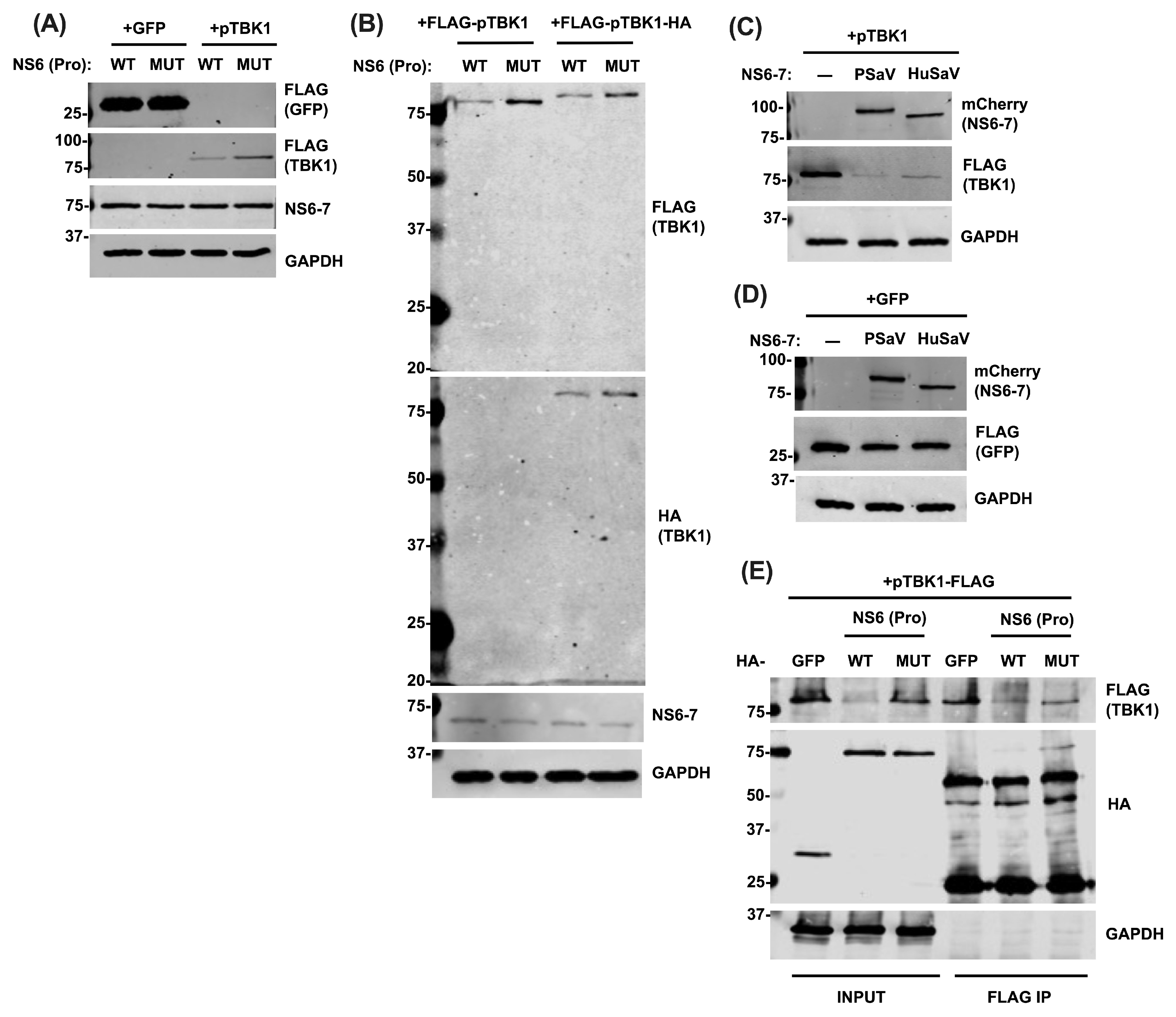
3.3. NS6 Interacts with and Modulates the Protein Levels of TBK1
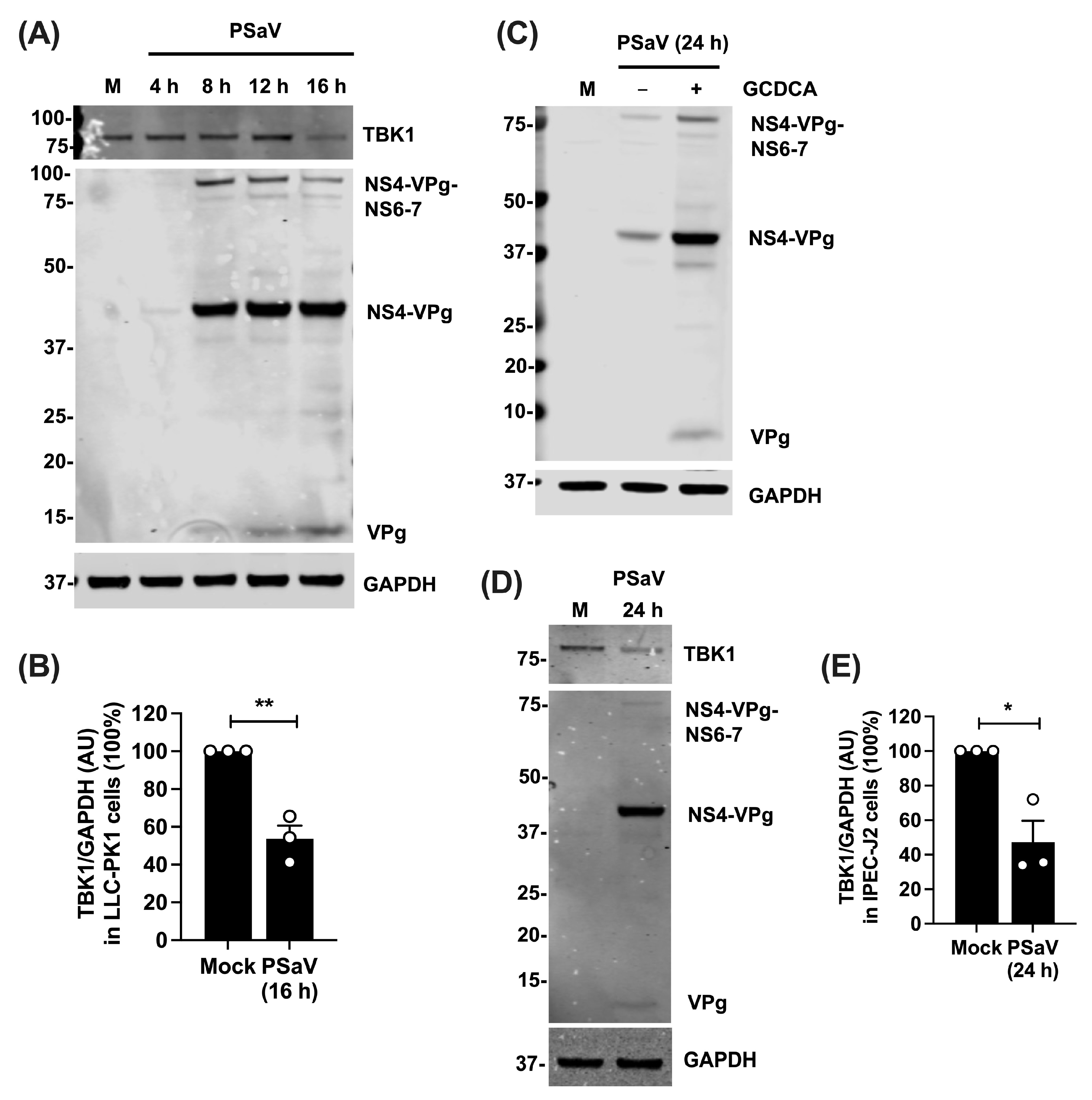
3.4. TBK1 Protein Levels Are Reduced during PSaV Infection
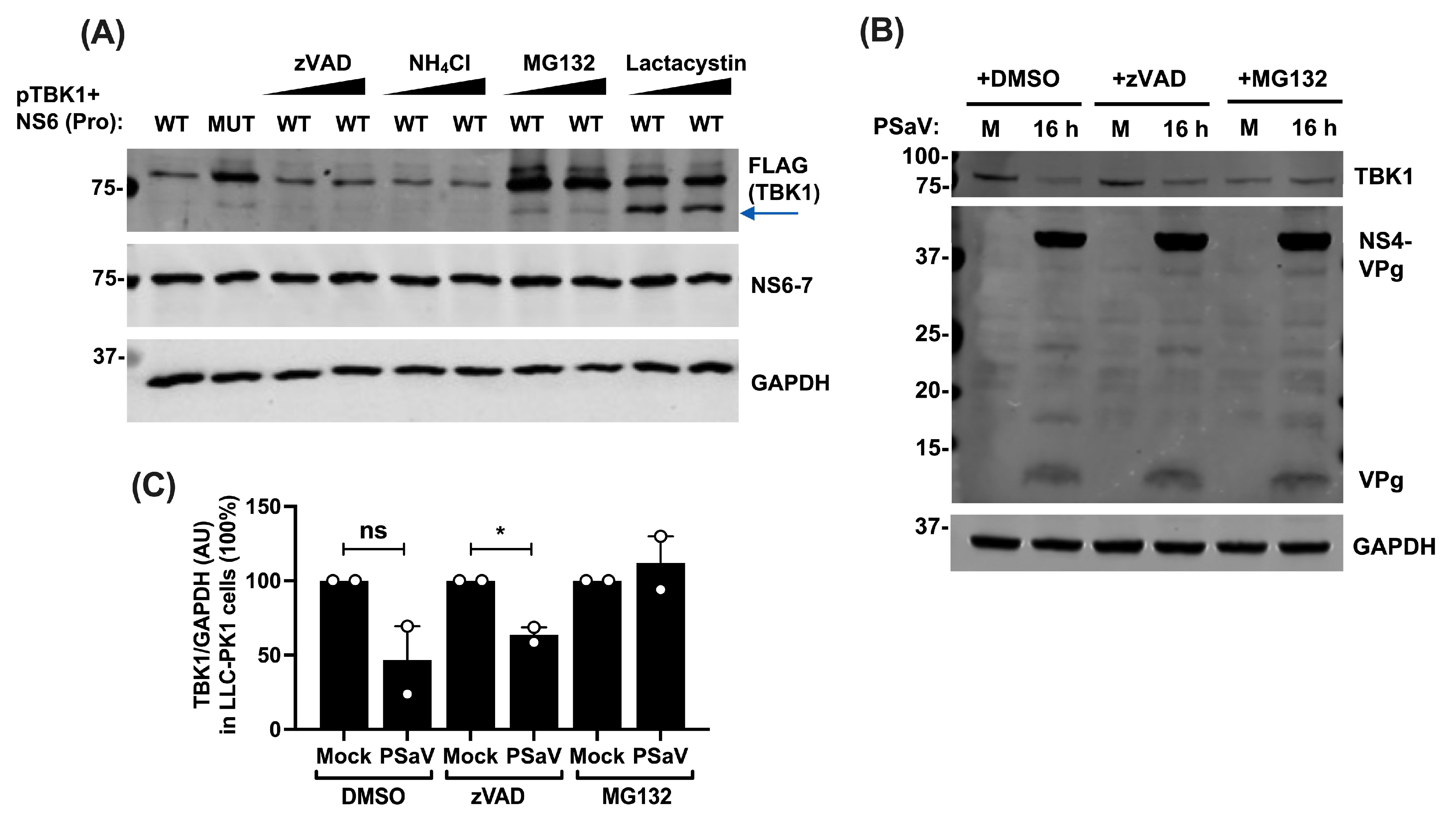
3.5. NS6-Mediated Reduction in TBK1 Levels Is Proteasomal-Dependent
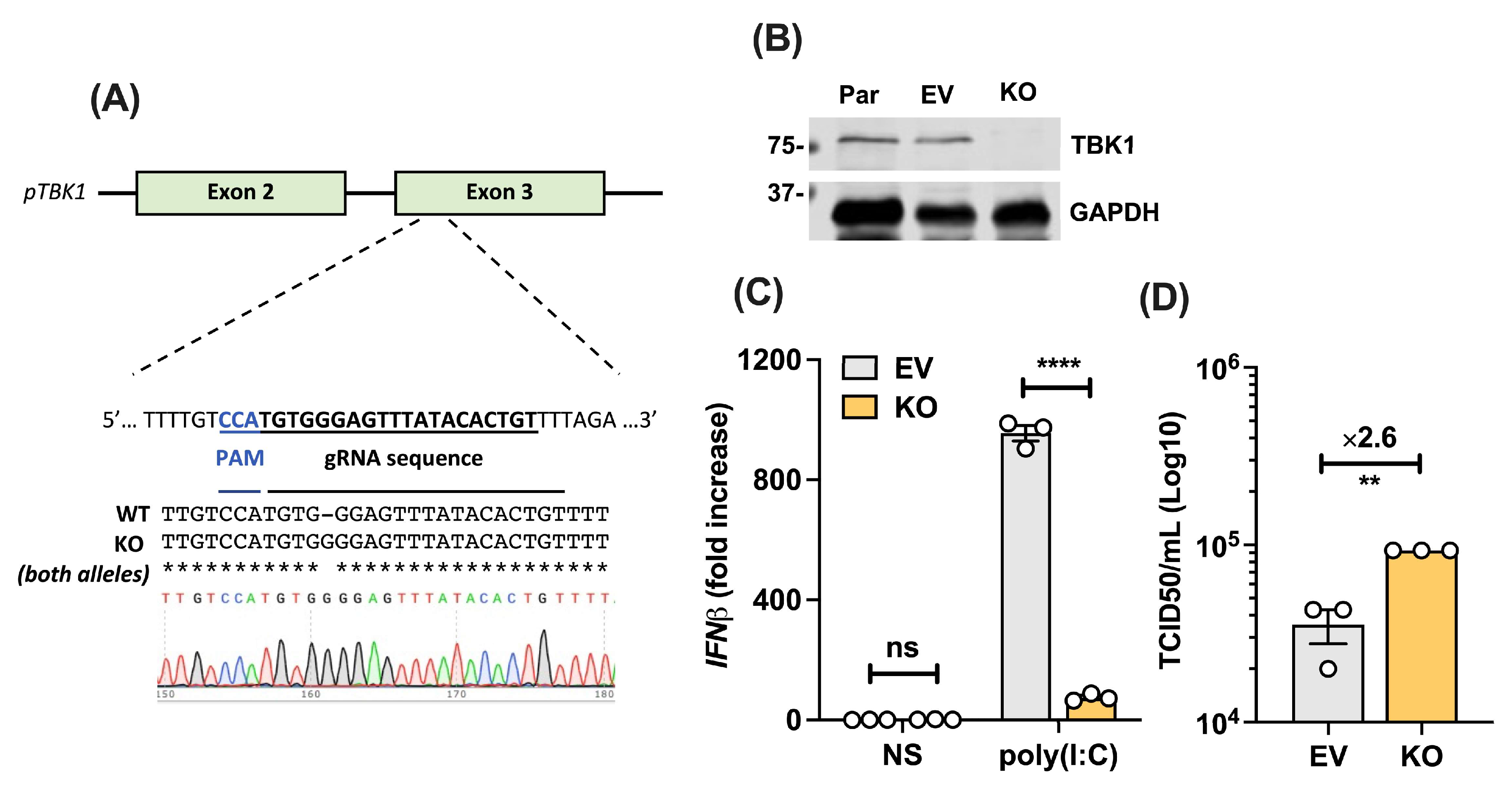
3.6. PSaV Infection in TBK1-Deficient Cells Results in Higher Viral Titres
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Zhang, W.; Cui, L.; Shen, Q.; Hua, X. Metagenomic identification, genetic characterization and genotyping of porcine sapoviruses. Infect. Genet. Evol. 2018, 62, 244–252. [Google Scholar] [CrossRef]
- Yinda, C.K.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Maes, P.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent sapoviruses detected by metagenomics analysis in straw-colored fruit bats in Cameroon. Emerg. Microbes Infect. 2017, 6, e38. [Google Scholar] [CrossRef]
- Oka, T.; Wang, Q.; Katayama, K.; Saif Linda, J. Comprehensive Review of Human Sapoviruses. Clin. Microbiol. Rev. 2015, 28, 32–53. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Katayama, K.; Ogawa, S.; Hansman, G.S.; Kageyama, T.; Ushijima, H.; Miyamura, T.; Takeda, N. Proteolytic Processing of Sapovirus ORF1 Polyprotein. J. Virol. 2005, 79, 7283–7290. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-C.; Kataoka, M.; Doan, Y.H.; Saito, H.; Takagi, H.; Muramatsu, M.; Oka, T. Characterization of a Human Sapovirus Genotype GII.3 Strain Generated by a Reverse Genetics System: VP2 Is a Minor Structural Protein of the Virion. Viruses 2022, 14, 1649. [Google Scholar] [CrossRef] [PubMed]
- Sosnovtseva, S.A.; Sosnovtsev, S.V.; Green, K.Y. Mapping of the feline calicivirus proteinase responsible for autocatalytic processing of the nonstructural polyprotein and identification of a stable proteinase-polymerase precursor protein. J. Virol. 1999, 73, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Sakuma, Y.; Kogasaka, R.; Akihara, M.; Horino, K.; Nakao, T.; Fukui, S. An outbreak of gastroenteritis associated with calicivirus in an infant home. J. Med. Virol. 1979, 4, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Madeley, C.R.; Cosgrove, B.P. CALICIVIRUSES IN MAN. Lancet 1976, 307, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Euller-Nicolas, G.; Le Mennec, C.; Schaeffer, J.; Zeng, X.-L.; Ettayebi, K.; Atmar Robert, L.; Le Guyader Françoise, S.; Estes Mary, K.; Desdouits, M. Human Sapovirus Replication in Human Intestinal Enteroids. J. Virol. 2023, 97, e00383-23. [Google Scholar] [CrossRef]
- Matsumoto, N.; Kurokawa, S.; Tamiya, S.; Nakamura, Y.; Sakon, N.; Okitsu, S.; Ushijima, H.; Yuki, Y.; Kiyono, H.; Sato, S. Replication of Human Sapovirus in Human-Induced Pluripotent Stem Cell-Derived Intestinal Epithelial Cells. Viruses 2023, 15, 1929. [Google Scholar] [CrossRef]
- Takagi, H.; Oka, T.; Shimoike, T.; Saito, H.; Kobayashi, T.; Takahashi, T.; Tatsumi, C.; Kataoka, M.; Wang, Q.; Saif, L.J.; et al. Human sapovirus propagation in human cell lines supplemented with bile acids. Proc. Natl. Acad. Sci. USA 2020, 117, 32078–32085. [Google Scholar] [CrossRef]
- Chang, K.-O.; Kim, Y.; Green, K.Y.; Saif, L.J. Cell-Culture Propagation of Porcine Enteric Calicivirus Mediated by Intestinal Contents Is Dependent on the Cyclic AMP Signaling Pathway. Virology 2002, 304, 302–310. [Google Scholar] [CrossRef]
- Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Kim, Y.; Saif, L.J.; Green, K.Y. Bile acids are essential for porcine enteric calicivirus replication in association with down-regulation of signal transducer and activator of transcription 1. Proc. Natl. Acad. Sci. USA 2004, 101, 8733–8738. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Wang, Q.; Saif, L.J.; Green, K.Y. Reverse genetics system for porcine enteric calicivirus, a prototype sapovirus in the Caliciviridae. J. Virol. 2005, 79, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Wang, Q.; Oka, T.; Saif, L.J. Porcine sapoviruses: Pathogenesis, epidemiology, genetic diversity, and diagnosis. Virus Res. 2020, 286, 198025. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Lorusso, E.; Banyai, K.; Decaro, N.; Corrente, M.; Elia, G.; Cavalli, A.; Radogna, A.; Costantini, V.; Saif, L.J.; et al. Identification of a porcine calicivirus related genetically to human sapoviruses. J. Clin. Microbiol. 2008, 46, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Jahun, A.S.; Goodfellow, I.G. Interferon responses to norovirus infections: Current and future perspectives. J. Gen. Virol. 2021, 102, 001660. [Google Scholar] [CrossRef] [PubMed]
- Hosmillo, M.; Sorgeloos, F.; Hiraide, R.; Lu, J.; Goodfellow, I.; Cho, K.O. Porcine sapovirus replication is restricted by the type I interferon response in cell culture. J. Gen. Virol. 2015, 96, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Smale, S.T. Selective Transcription in Response to an Inflammatory Stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef]
- Berschneider, H. Development of normal cultured small intestinal epithelial cell lines which transport Na and Cl. (Abstract). Gasteroenterology 1989, 96, A41. [Google Scholar]
- Geens, M.M.; Niewold, T.A. Optimizing culture conditions of a porcine epithelial cell line IPEC-J2 through a histological and physiological characterization. Cytotechnology 2011, 63, 415–423. [Google Scholar] [CrossRef]
- Odon, V.; Georgana, I.; Holley, J.; Morata, J.; Maluquer de Motes, C. Novel Class of Viral Ankyrin Proteins Targeting the Host E3 Ubiquitin Ligase Cullin-2. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-Dependent Phosphorylation of the IRF-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Ranjith-Kumar, C.T.; Wen, Y.; Baxter, N.; Bhardwaj, K.; Cheng Kao, C. A Cell-Based Assay for RNA Synthesis by the HCV Polymerase Reveals New Insights on Mechanism of Polymerase Inhibitors and Modulation by NS5A. PLoS ONE 2011, 6, e22575. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- REED, L.J.; Muench, H. A Simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Lei, J.; Hilgenfeld, R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, P.; Chen, Z.; Wang, D.; Zhou, Y.; Zhu, X.; Xiao, S.; Fang, L. Norovirus 3C-Like protease antagonizes interferon-β production by cleaving NEMO. Virology 2022, 571, 12–20. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Oka, T.; Yamamoto, M.; Katayama, K.; Hansman, G.S.; Ogawa, S.; Miyamura, T.; Takeda, N. Identification of the cleavage sites of sapovirus open reading frame 1 polyprotein. J. Gen. Virol. 2006, 87, 3329–3338. [Google Scholar] [CrossRef]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z.; et al. Foot-and-Mouth Disease Virus 3C Protease Cleaves NEMO To Impair Innate Immune Signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef] [PubMed]
- Men, Y.; Wang, Y.; Wang, H.; Zhang, M.; Liu, J.; Chen, Y.; Han, X.; Chen, R.; Chen, Q.; Hu, A. RHDV 3C protein antagonizes type I interferon signaling by cleaving interferon promoter stimulated 1 protein. Virus Genes 2023, 59, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Weidman, M.K.; Yalamanchili, P.; Ng, B.; Tsai, W.; Dasgupta, A. Poliovirus 3C Protease-Mediated Degradation of Transcriptional Activator p53 Requires a Cellular Activity. Virology 2001, 291, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, H.; Zhang, X.; Li, K.; Yang, F.; Cao, W.; Liu, H.; Gao, L.; Xue, Z.; Liu, X.; et al. Porcine Picornavirus 3C Protease Degrades PRDX6 to Impair PRDX6-mediated Antiviral Function. Virol. Sin. 2021, 36, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Jeremiah, S.S.; Miyakawa, K.; Matsunaga, S.; Nishi, M.; Kudoh, A.; Takaoka, A.; Sawasaki, T.; Ryo, A. Cleavage of TANK-Binding Kinase 1 by HIV-1 Protease Triggers Viral Innate Immune Evasion. Front. Microbiol. 2021, 12, 643407. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Lin, X.-T.; Zhao, S.; Zheng, X.-Q.; Zhou, Y.-Q.; Xiao, L.-L.; Chen, H.; Zhang, Z.-Y.; Zhang, L.-J.; Wu, X.-X. Tripartite motif-containing protein 46 accelerates influenza A H7N9 virus infection by promoting K48-linked ubiquitination of TBK1. Virol. J. 2022, 19, 176. [Google Scholar] [CrossRef]
- Sawaged, S.; Mota, T.; Piplani, H.; Thakur, R.; Lall, D.; McCabe, E.; Seo, S.; Sutterwala, F.S.; Feuer, R.; Gottlieb, R.A.; et al. TBK1 and GABARAP family members suppress Coxsackievirus B infection by limiting viral production and promoting autophagic degradation of viral extracellular vesicles. PLoS Pathog. 2022, 18, e1010350. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, I.; Volety, I.; Shukla, D. OPTN-TBK1 axis and a role for PLK1 in HSV-1 infection. mBio 2023, 14, e02715-23. [Google Scholar] [CrossRef] [PubMed]
- Jahun, A.S.; Sorgeloos, F.; Chaudhry, Y.; Arthur, S.E.; Hosmillo, M.; Georgana, I.; Izuagbe, R.; Goodfellow, I.G. Leaked genomic and mitochondrial DNA contribute to the host response to noroviruses in a STING-dependent manner. Cell Rep. 2023, 42, 112179. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Devos, J.M.; Ng, S.-L.; Nanao, M.H.; Round, A.; Maniatis, T.; Panne, D. Crystal Structure and Mechanism of Activation of TANK-Binding Kinase 1. Cell Rep. 2013, 3, 734–746. [Google Scholar] [CrossRef]
- Sharif, M.; Baek, Y.-B.; Nguyen, T.H.; Soliman, M.; Cho, K.-O. Porcine sapovirus-induced RIPK1-dependent necroptosis is proviral in LLC-PK cells. PLoS ONE 2023, 18, e0279843. [Google Scholar] [CrossRef]
- Delanghe, T.; Dondelinger, Y.; Bertrand, M.J.M. RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends Cell Biol. 2020, 30, 189–200. [Google Scholar] [CrossRef]
- Schierack, P.; Nordhoff, M.; Pollmann, M.; Weyrauch, K.D.; Amasheh, S.; Lodemann, U.; Jores, J.; Tachu, B.; Kleta, S.; Blikslager, A.; et al. Characterization of a porcine intestinal epithelial cell line for in vitro studies of microbial pathogenesis in swine. Histochem. Cell Biol. 2006, 125, 293–305. [Google Scholar] [CrossRef]
- Brosnahan, A.J.; Brown, D.R. Porcine IPEC-J2 intestinal epithelial cells in microbiological investigations. Vet. Microbiol. 2012, 156, 229–237. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgana, I.; Hosmillo, M.; Jahun, A.S.; Emmott, E.; Sorgeloos, F.; Cho, K.-O.; Goodfellow, I.G. Porcine Sapovirus Protease Controls the Innate Immune Response and Targets TBK1. Viruses 2024, 16, 247. https://doi.org/10.3390/v16020247
Georgana I, Hosmillo M, Jahun AS, Emmott E, Sorgeloos F, Cho K-O, Goodfellow IG. Porcine Sapovirus Protease Controls the Innate Immune Response and Targets TBK1. Viruses. 2024; 16(2):247. https://doi.org/10.3390/v16020247
Chicago/Turabian StyleGeorgana, Iliana, Myra Hosmillo, Aminu S. Jahun, Edward Emmott, Frederic Sorgeloos, Kyoung-Oh Cho, and Ian G. Goodfellow. 2024. "Porcine Sapovirus Protease Controls the Innate Immune Response and Targets TBK1" Viruses 16, no. 2: 247. https://doi.org/10.3390/v16020247
APA StyleGeorgana, I., Hosmillo, M., Jahun, A. S., Emmott, E., Sorgeloos, F., Cho, K. -O., & Goodfellow, I. G. (2024). Porcine Sapovirus Protease Controls the Innate Immune Response and Targets TBK1. Viruses, 16(2), 247. https://doi.org/10.3390/v16020247