
Genetics of H5N1 and H5N8 High-Pathogenicity Avian Influenza Viruses Isolated in Japan in Winter 2021–2022
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation and Whole-Genome Sequencing
2.2. Phylogenetic Analysis
3. Results
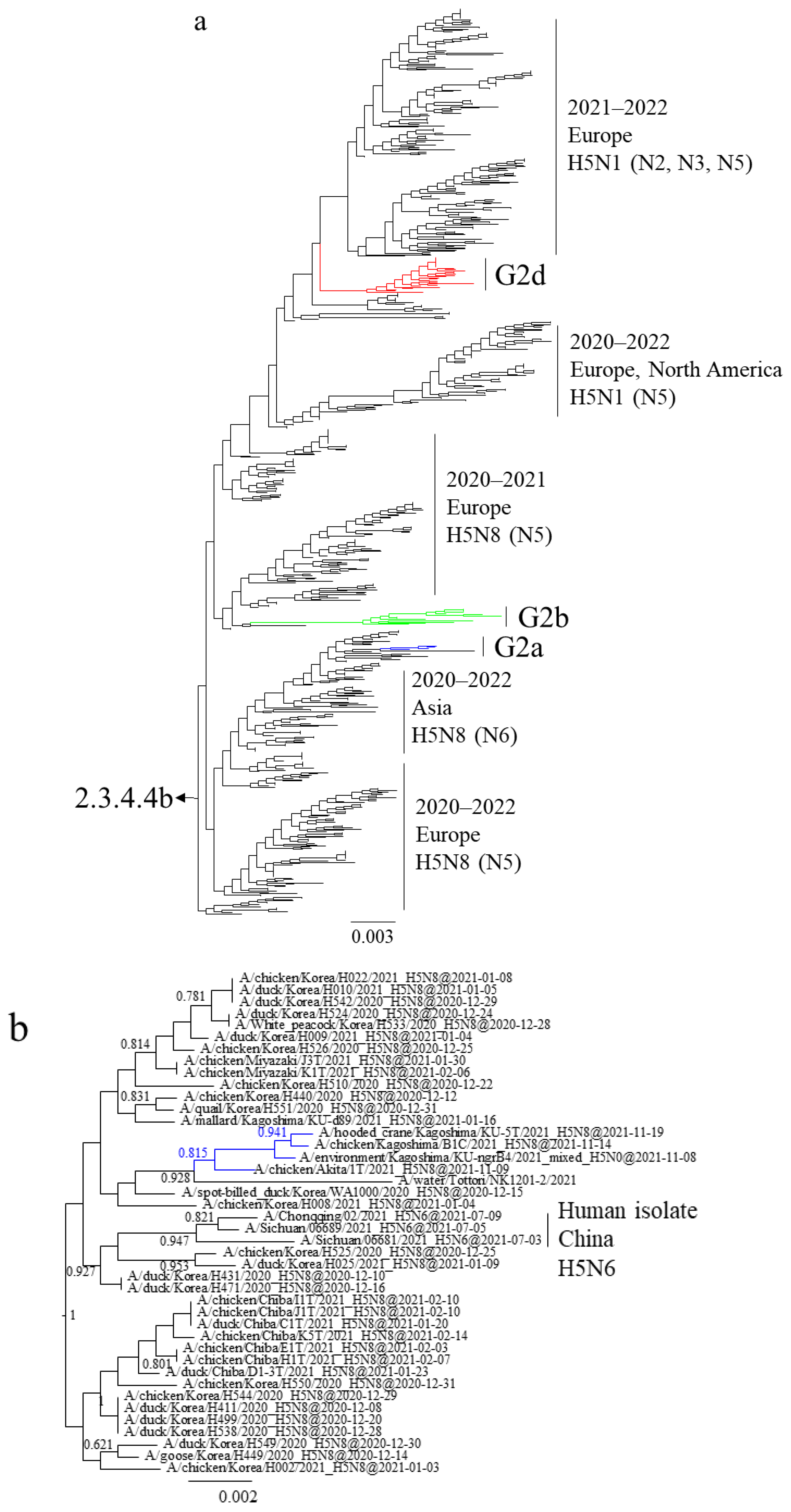
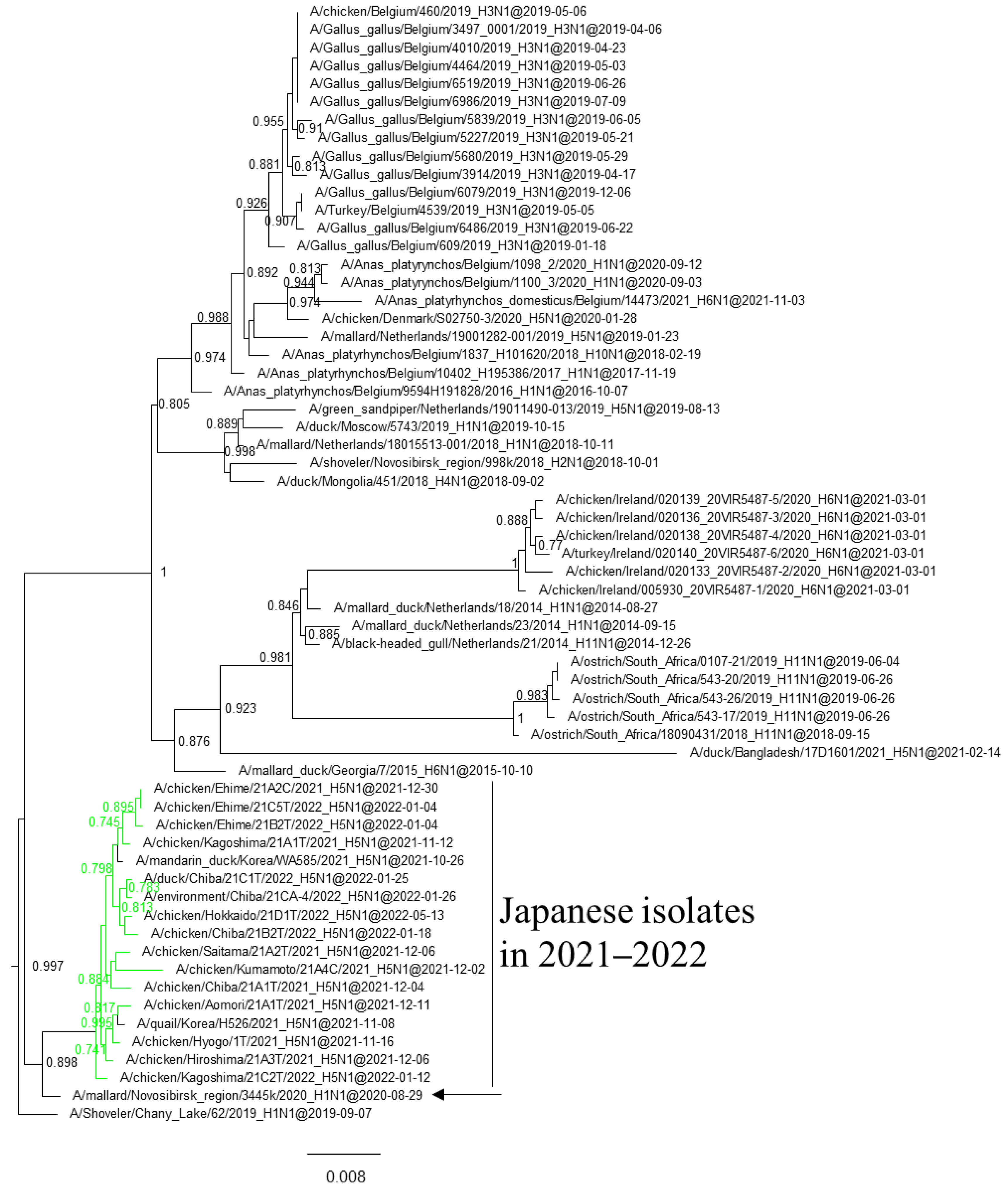
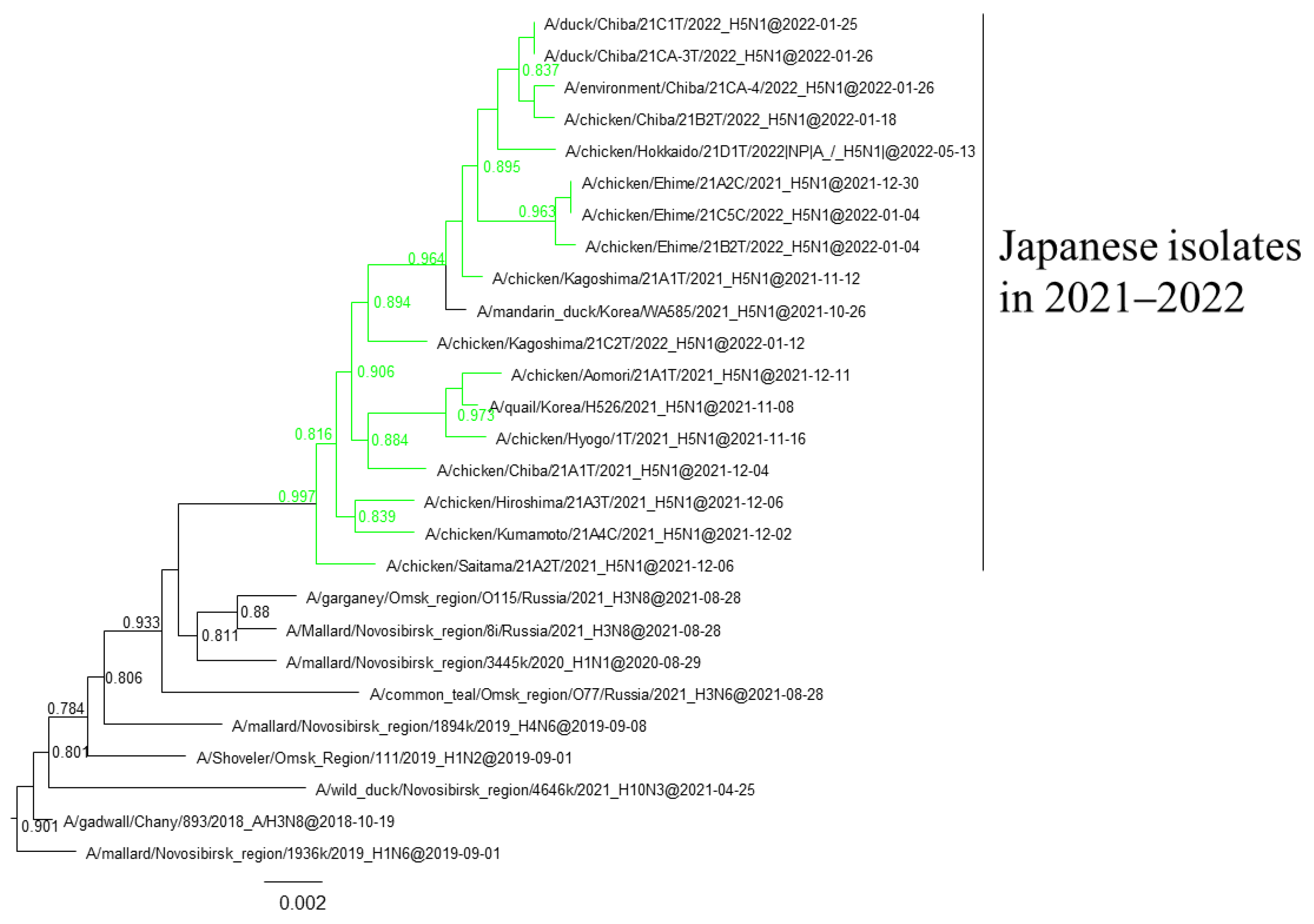
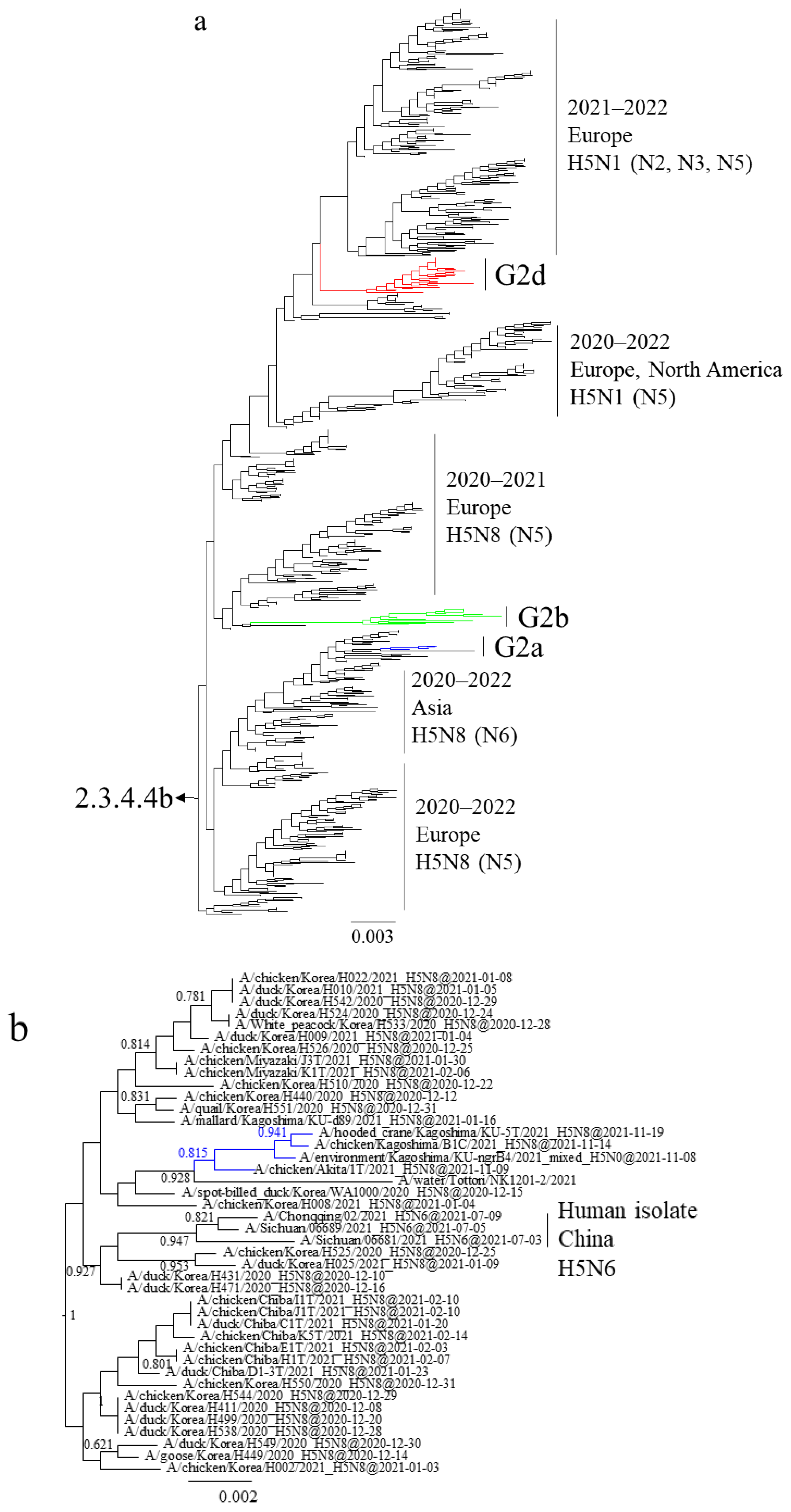
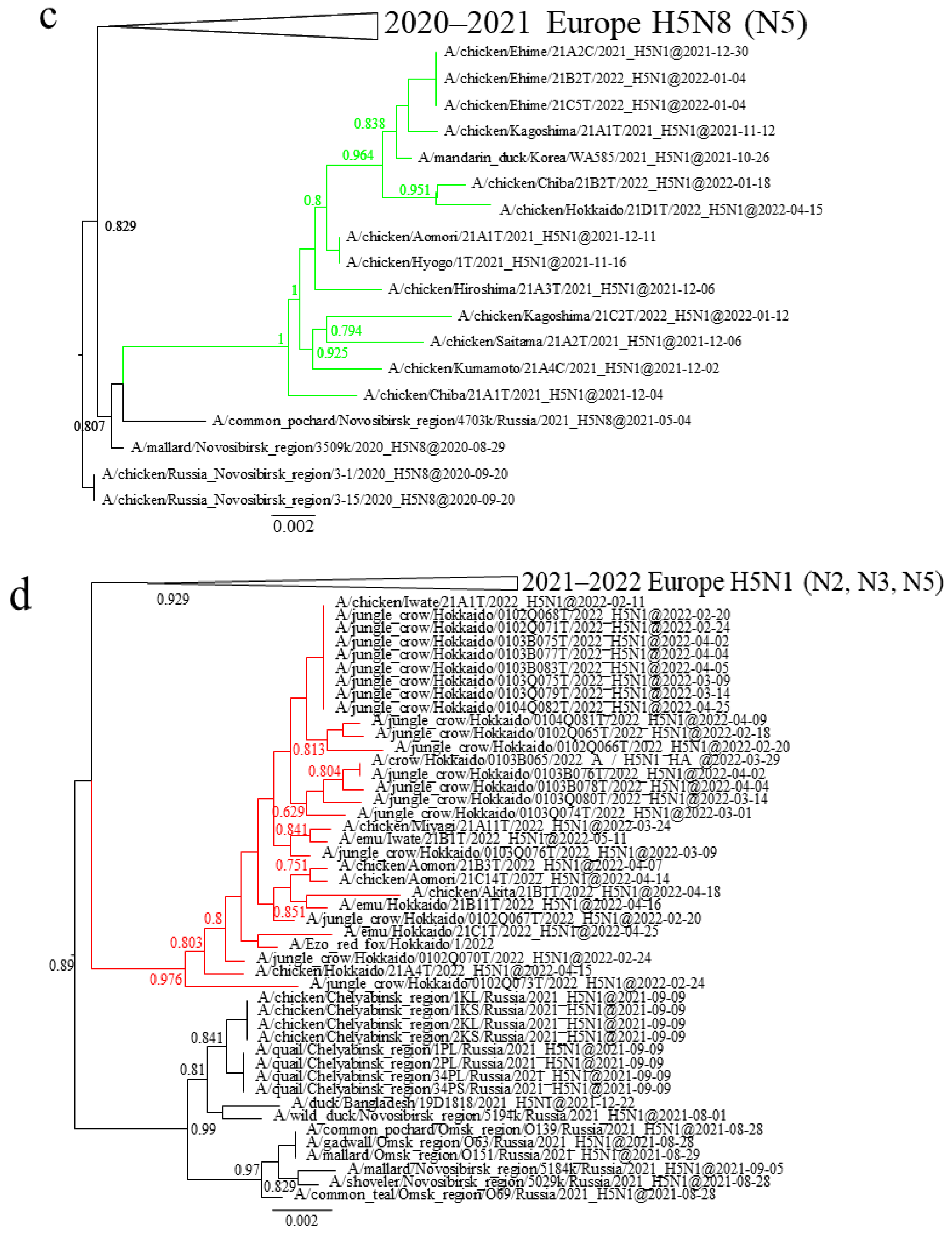
3.1. Phylogenetic Analysis of Japanese H5 Isolates from the 2021–2022 Winter
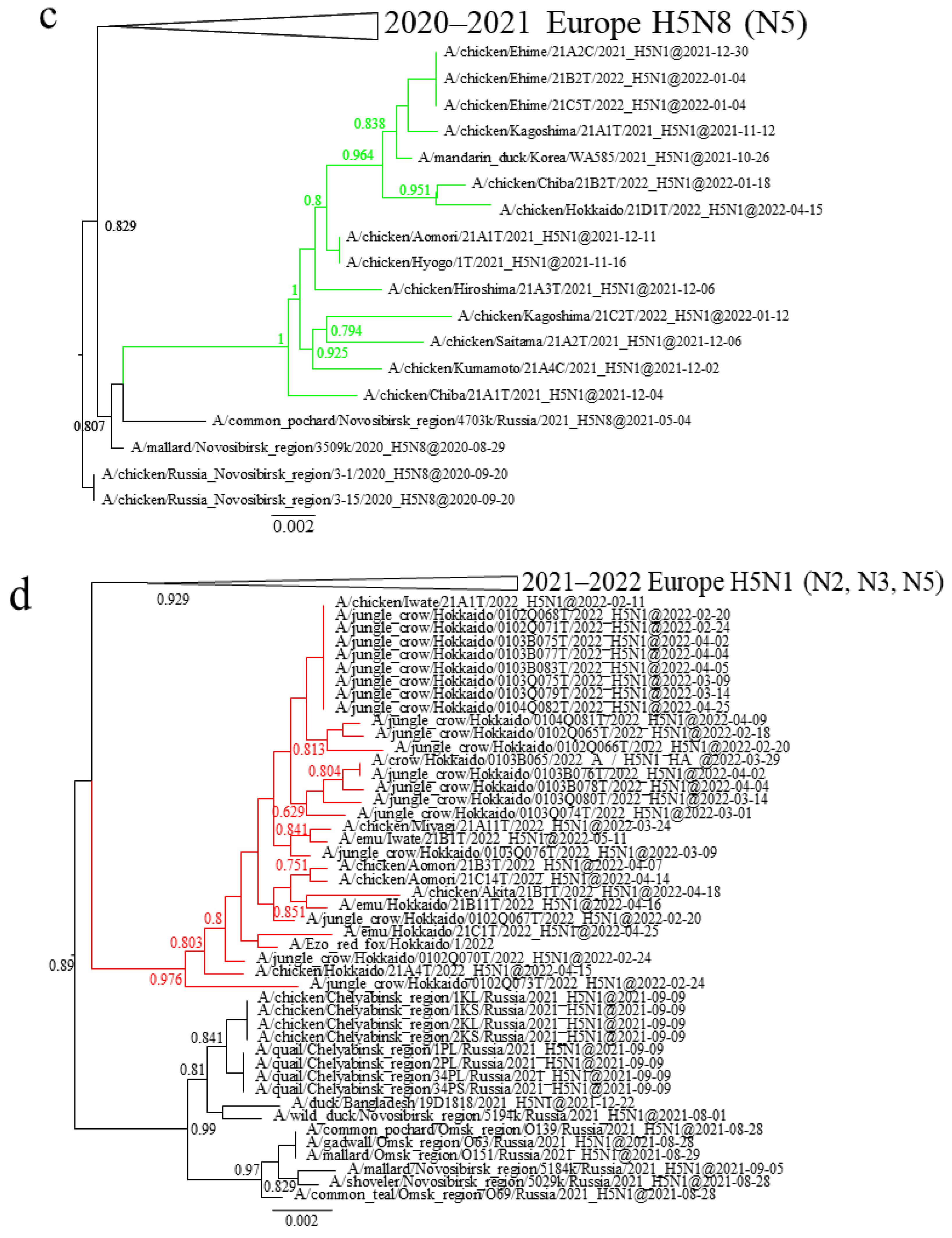
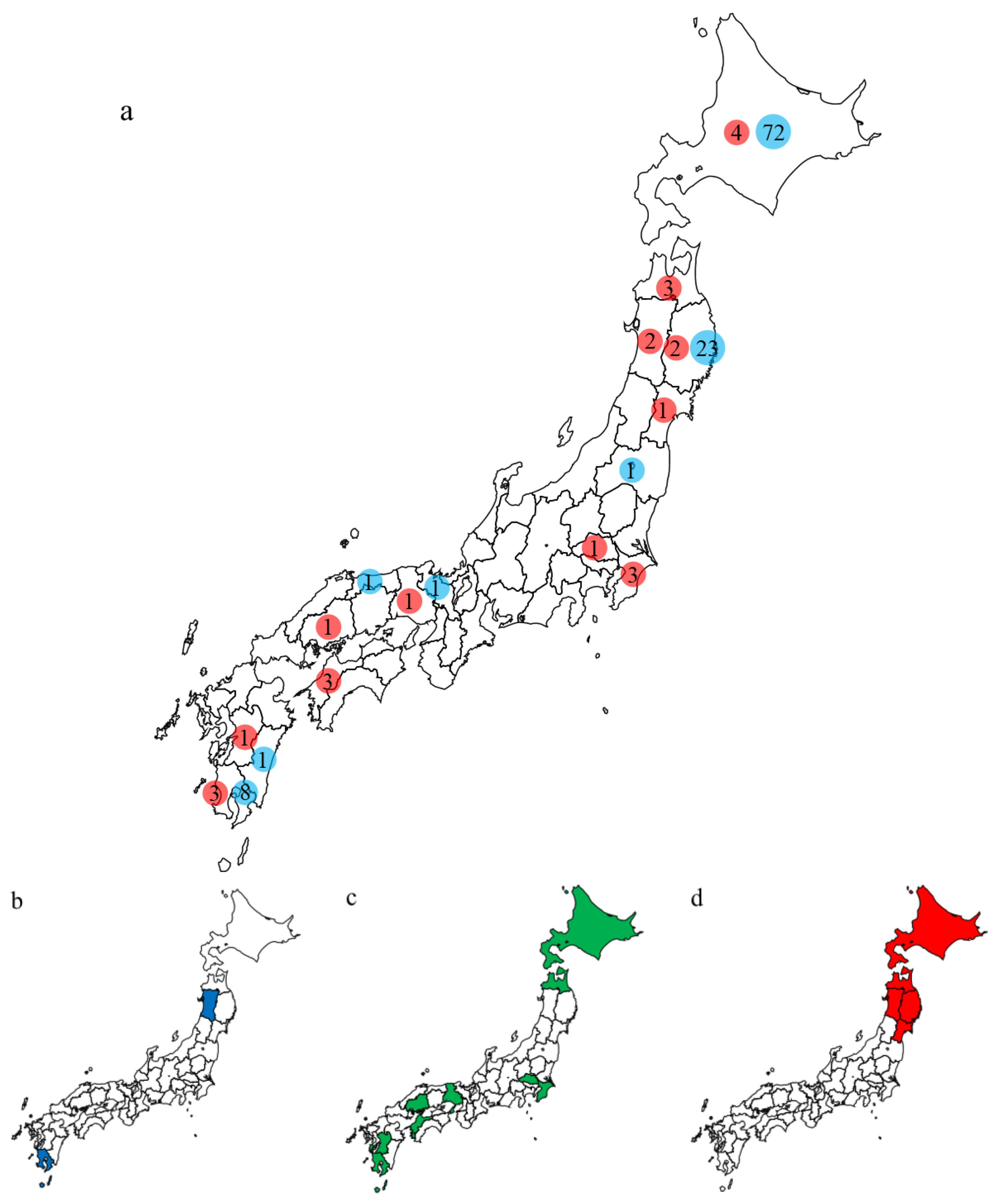
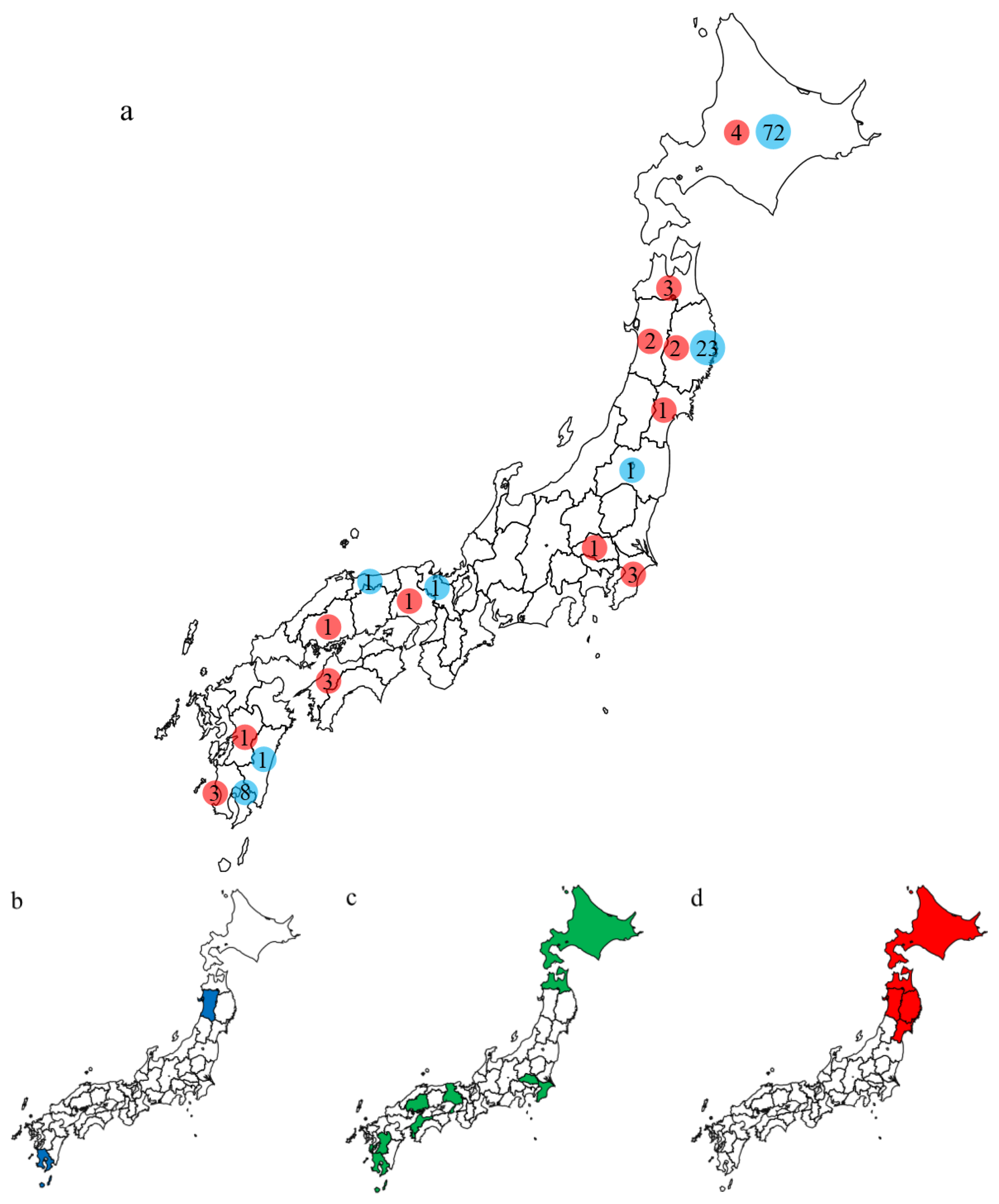
3.2. Characteristics of the Outbreaks in Japan during 2021–2022
3.3. Gene Constellations of H5 HPAIVs Isolated in Japan in the 2021–2022 Winter
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Criado, M.F.; Swayne, D.E. Pathobiological Origins and Evolutionary History of Highly Pathogenic Avian Influenza Viruses. Cold Spring Harb. Perspect. Med. 2021, 11, a038679. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.D.; Donis, R.O.; World Health Organization/World Organisation for Animal Health/Food and Agriculture Organization (WHO/OIE/FAO) H5 Evolution Working Group. Nomenclature updates resulting from the evolution of avian influenza A(H5) virus clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013–2014. Influenza Other Respir. Viruses 2015, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef]
- Lycett, S.J.; Duchatel, F.; Digard, P. A brief history of bird flu. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180257. [Google Scholar] [CrossRef]
- Mase, M.; Tsukamoto, K.; Imada, T.; Imai, K.; Tanimura, N.; Nakamura, K.; Yamamoto, Y.; Hitomi, T.; Kira, T.; Nakai, T.; et al. Characterization of H5N1 influenza A viruses isolated during the 2003–2004 influenza outbreaks in Japan. Virology 2005, 332, 167–176. [Google Scholar] [CrossRef]
- Uchida, Y.; Mase, M.; Yoneda, K.; Kimura, A.; Obara, T.; Kumagai, S.; Saito, T.; Yamamoto, Y.; Nakamura, K.; Tsukamoto, K.; et al. Highly pathogenic avian influenza virus (H5N1) isolated from whooper swans, Japan. Emerg. Infect. Dis. 2008, 14, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Shivakoti, S.; Ito, H.; Otsuki, K.; Ito, T. Characterization of H5N1 highly pathogenic avian influenza virus isolated from a mountain hawk eagle in Japan. J. Vet. Med. Sci. 2010, 72, 459–463. [Google Scholar] [CrossRef]
- Uchida, Y.; Suzuki, Y.; Shirakura, M.; Kawaguchi, A.; Nobusawa, E.; Tanikawa, T.; Hikono, H.; Takemae, N.; Mase, M.; Kanehira, K.; et al. Genetics and infectivity of H5N1 highly pathogenic avian influenza viruses isolated from chickens and wild birds in Japan during 2010–2011. Virus Res. 2012, 170, 109–117. [Google Scholar] [CrossRef]
- Sakoda, Y.; Ito, H.; Uchida, Y.; Okamatsu, M.; Yamamoto, N.; Soda, K.; Nomura, N.; Kuribayashi, S.; Shichinohe, S.; Sunden, Y.; et al. Reintroduction of H5N1 highly pathogenic avian influenza virus by migratory water birds, causing poultry outbreaks in the 2010–2011 winter season in Japan. J. Gen. Virol. 2012, 93 Pt 3, 541–550. [Google Scholar] [CrossRef]
- Kanehira, K.; Uchida, Y.; Takemae, N.; Hikono, H.; Tsunekuni, R.; Saito, T. Characterization of an H5N8 influenza A virus isolated from chickens during an outbreak of severe avian influenza in Japan in April 2014. Arch. Virol. 2015, 160, 1629–1643. [Google Scholar] [CrossRef]
- Tanikawa, T.; Kanehira, K.; Tsunekuni, R.; Uchida, Y.; Takemae, N.; Saito, T. Pathogenicity of H5N8 highly pathogenic avian influenza viruses isolated from a wild bird fecal specimen and a chicken in Japan in 2014. Microbiol. Immunol. 2016, 60, 243–252. [Google Scholar] [CrossRef]
- Takemae, N.; Tsunekuni, R.; Sharshov, K.; Tanikawa, T.; Uchida, Y.; Ito, H.; Soda, K.; Usui, T.; Sobolev, I.; Shestopalov, A.; et al. Five distinct reassortants of H5N6 highly pathogenic avian influenza A viruses affected Japan during the winter of 2016–2017. Virology 2017, 512, 8–20. [Google Scholar] [CrossRef]
- Tsunekuni, R.; Yaguchi, Y.; Kashima, Y.; Yamashita, K.; Takemae, N.; Mine, J.; Tanikawa, T.; Uchida, Y.; Saito, T. Spatial transmission of H5N6 highly pathogenic avian influenza viruses among wild birds in Ibaraki Prefecture, Japan, 2016–2017. Arch. Virol. 2018, 163, 1195–1207. [Google Scholar] [CrossRef]
- Ozawa, M.; Matsuu, A.; Khalil, A.M.; Nishi, N.; Tokorozaki, K.; Masatani, T.; Horie, M.; Okuya, K.; Ueno, K.; Kuwahara, M.; et al. Phylogenetic variations of highly pathogenic H5N6 avian influenza viruses isolated from wild birds in the Izumi plain, Japan, during the 2016–2017 winter season. Transbound. Emerg. Dis. 2018, 66, 797–806. [Google Scholar] [CrossRef]
- Mine, J.; Uchida, Y.; Nakayama, M.; Tanikawa, T.; Tsunekuni, R.; Sharshov, K.; Takemae, N.; Sobolev, I.; Shestpalov, A.; Saito, T. Genetics and pathogenicity of H5N6 highly pathogenic avian influenza viruses isolated from wild birds and a chicken in Japan during winter 2017–2018. Virology 2019, 533, 1–11. [Google Scholar] [CrossRef]
- Mine, J.; Tsunekuni, R.; Tanikawa, T.; Uchida, Y.; Dubovitskiy, N.; Derko, A.; Sobolev, I.; Shestopalov, A.; Sharshov, K.; Saito, T. Genetics of Japanese H5N8 high pathogenicity avian influenza viruses isolated in winter 2020–2021 and their genetic relationship with avian influenza viruses in Siberia. Transbound. Emerg. Dis. 2022, 69, e2195–e2213. [Google Scholar] [CrossRef]
- Baek, Y.G.; Lee, Y.N.; Lee, D.H.; Shin, J.I.; Lee, J.H.; Chung, D.H.; Lee, E.K.; Heo, G.B.; Sagong, M.; Kye, S.J.; et al. Multiple reassortants of H5N8 Clade 2.3.4.4b Highly pathogenic avian influenza viruses detected in South Korea during the winter of 2020–2021. Viruses 2021, 13, 490. [Google Scholar] [CrossRef]
- Okuya, K.; Mine, J.; Tokorozaki, K.; Kojima, I.; Esaki, M.; Miyazawa, K.; Tsunekuni, R.; Sakuma, S.; Kumagai, A.; Takadate, Y.; et al. Genetically diverse highly pathogenic avian influenza A (H5N1/H5N8) viruses among wild waterfowl and domestic poultry, Japan, 2021. Emerg. Infect. Dis. 2022, 28, 1451–1455. [Google Scholar] [CrossRef]
- Isoda, N.; Onuma, M.; Hiono, T.; Sobolev, I.; Lim, H.Y.; Nabeshima, K.; Honjyo, H.; Yokoyama, M.; Shestopalov, A.; Sakoda, Y. Detection of New H5N1 High Pathogenicity avian influenza viruses in winter 2021–2022 in the Far East, which are genetically close to those in Europe. Viruses 2022, 14, 2168. [Google Scholar] [CrossRef]
- Takadate, Y.; Tsunekuni, R.; Kumagai, A.; Mine, J.; Kikutani, Y.; Sakuma, S.; Miyazawa, K.; Uchida, Y. Different Infectivity and Transmissibility of H5N8 and H5N1 High pathogenicity avian influenza viruses isolated from chickens in Japan in the 2021/2022 season. Viruses 2023, 15, 265. [Google Scholar] [CrossRef]
- Soda, K.; Tomioka, Y.; Usui, T.; Ozaki, H.; Ito, H.; Nagai, Y.; Yamamoto, N.; Okamatsu, M.; Isoda, N.; Kajihara, M.; et al. Susceptibility of common dabbling and diving duck species to clade 2.3.2.1 H5N1 high pathogenicity avian influenza virus: An experimental infection study. J. Vet. Med. Sci. 2023, 85, 942–949. [Google Scholar] [CrossRef]
- Mine, J.; Uchida, Y.; Takemae, N.; Saito, T. Genetic Characterization of Influenza A Viruses in Japanese Swine in 2015 to 2019. J. Virol. 2020, 94, e02169-19. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Boere, G.C.; Stroud, D.A. The Flyway Concept: What it is and what it isn’t. In Waterbirds around the World; Boere, G.C., Galbraith, C.A., Stroud, D.A., Eds.; Stationery Office: Edinburgh, UK, 2006; pp. 40–47. [Google Scholar]
- Onuma, M.; Kakogawa, M.; Yanagisawa, M.; Haga, A.; Okano, T.; Neagari, Y.; Okano, T.; Goka, K.; Asakawa, M. Characterizing the temporal patterns of avian influenza virus introduction into Japan by migratory birds. J. Vet. Med. Sci. 2017, 79, 943–951. [Google Scholar] [CrossRef]
- Isoda, N.; Twabela, A.T.; Bazarragchaa, E.; Ogasawara, K.; Hayashi, H.; Wang, Z.J.; Kobayashi, D.; Watanabe, Y.; Saito, K.; Kida, H.; et al. Re-Invasion of H5N8 High Pathogenicity Avian Influenza Virus Clade 2.3.4.4b in Hokkaido, Japan, 2020. Viruses 2020, 12, 1439. [Google Scholar] [CrossRef]
- Khalil, A.M.; Fujimoto, Y.; Kojima, I.; Esaki, M.; Ri, K.; Masatani, T.; Matsui, T.; Ozawa, M. Genetic characterization of H5N8 highly pathogenic avian influenza viruses isolated from falcated ducks and environmental water in Japan in November 2020. Pathogens 2021, 10, 171. [Google Scholar] [CrossRef]
- Sakuma, S.; Uchida, Y.; Kajita, M.; Tanikawa, T.; Mine, J.; Tsunekuni, R.; Saito, T. First outbreak of an H5N8 highly pathogenic avian influenza virus on a chicken farm in Japan in 2020. Viruses 2021, 13, 489. [Google Scholar] [CrossRef]
- Sivay, M.V.; Sayfutdinova, S.G.; Sharshov, K.A.; Alekseev, A.Y.; Yurlov, A.K.; Runstadler, J.; Shestopalov, A.M. Surveillance of influenza A virus in wild birds in the Asian portion of Russia in 2008. Avian Dis. 2012, 56, 456–463. [Google Scholar] [CrossRef]
- Sharshov, K.; Sivay, M.; Liu, D.; Pantin-Jackwood, M.; Marchenko, V.; Durymanov, A.; Alekseev, A.; Damdindorj, T.; Gao, G.F.; Swayne, D.E.; et al. Molecular characterization and phylogenetics of a reassortant H13N8 influenza virus isolated from gulls in Mongolia. Virus Genes 2014, 49, 237–249. [Google Scholar] [CrossRef]
- Hatta, M.; Gao, P.; Halfmann, P.; Kawaoka, Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 2001, 293, 1840–1842. [Google Scholar] [CrossRef]
- Salomon, R.; Franks, J.; Govorkova, E.A.; Ilyushina, N.A.; Yen, H.L.; Hulse-Post, D.J.; Humberd, J.; Trichet, M.; Rehg, J.E.; Webby, R.J.; et al. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J. Exp. Med. 2006, 203, 689–697. [Google Scholar] [CrossRef]
- Subbarao, E.K.; London, W.; Murphy, B.R. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 1993, 67, 1761–1764. [Google Scholar] [CrossRef]
- Chen, G.W.; Chang, S.C.; Mok, C.K.; Lo, Y.L.; Kung, Y.N.; Huang, J.H.; Shih, Y.H.; Wang, J.Y.; Chiang, C.; Chen, C.J.; et al. Genomic signatures of human versus avian influenza A viruses. Emerg. Infect. Dis. 2006, 12, 1353–1360. [Google Scholar] [CrossRef]
- Bisset, A.T.; Hoyne, G.F. An outbreak of highly pathogenic avian influenza (H7N7) in Australia and the potential for novel influenza A viruses to emerge. Microorganisms 2021, 9, 1639. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
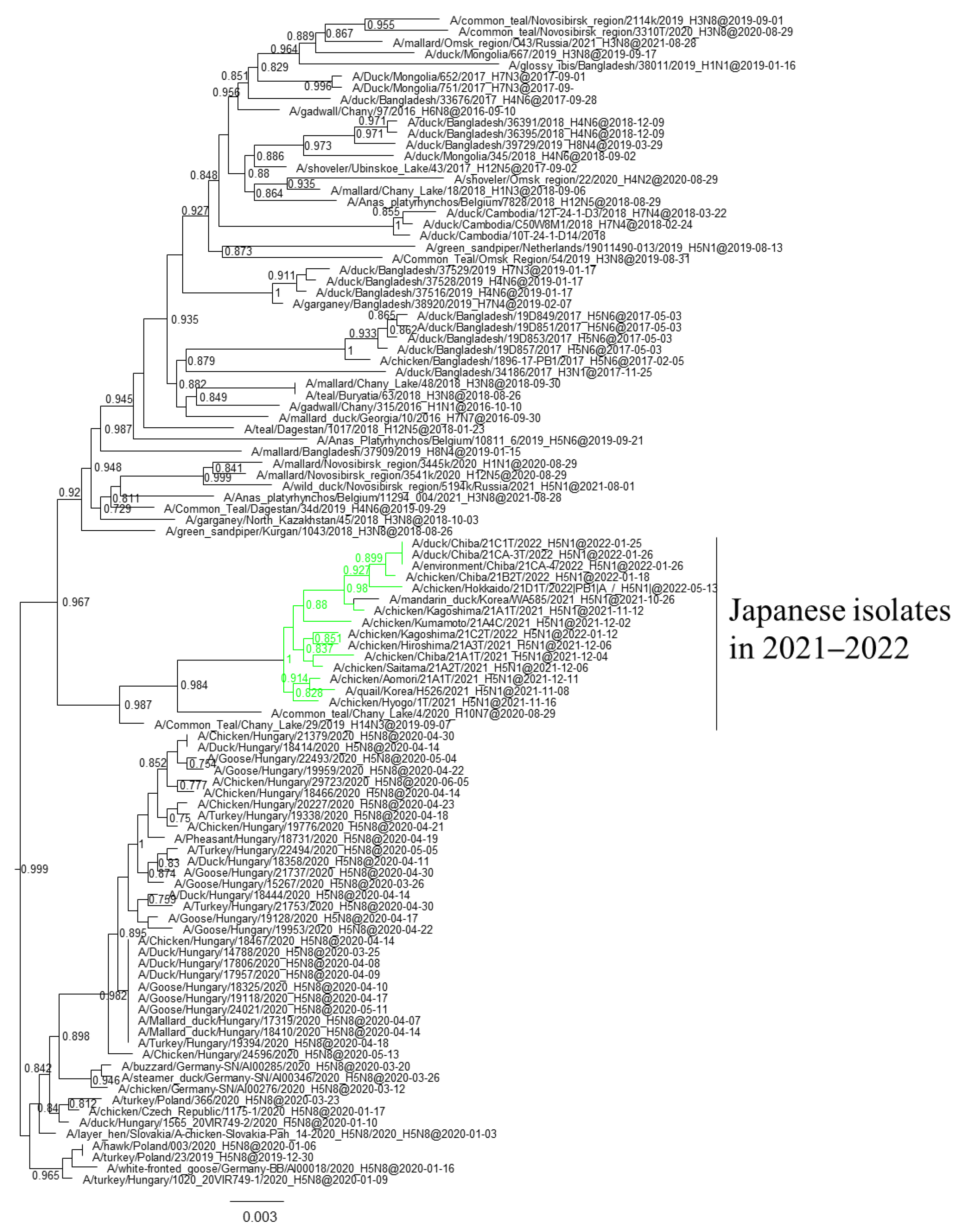{kind=link}
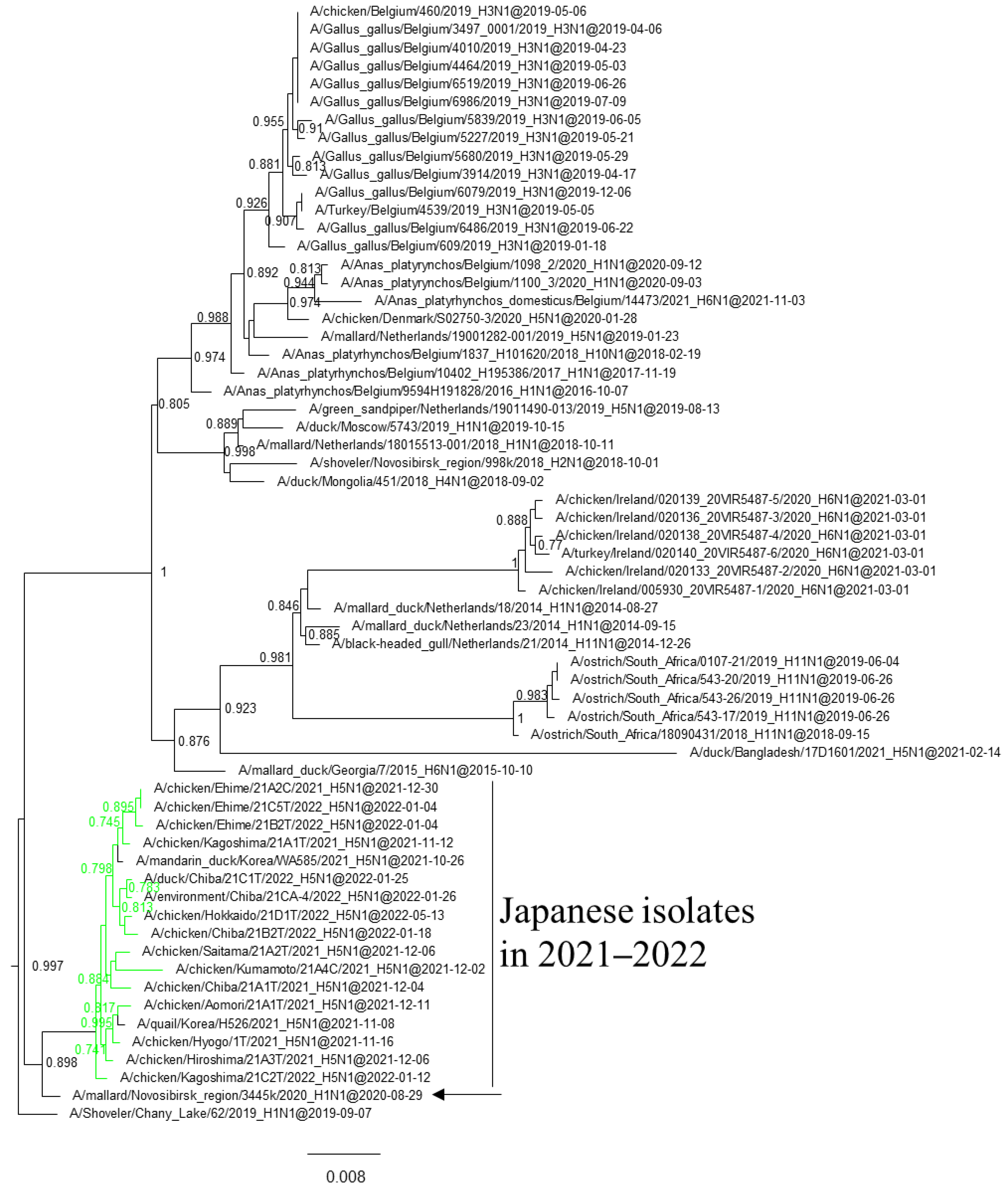{kind=link}
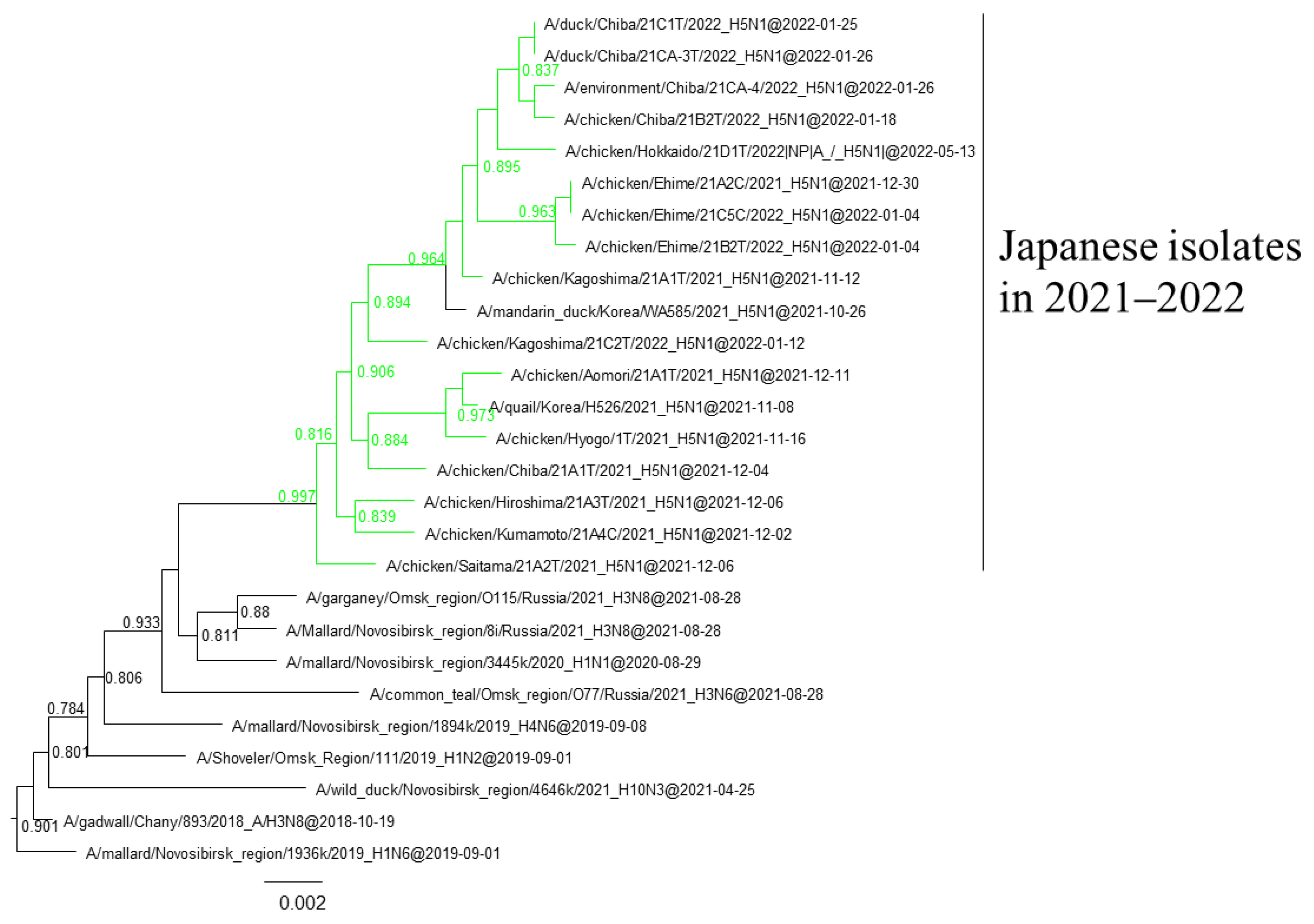{kind=link}
| Genotype | Collection Date | Host | Prefecture | Area | Subtype | Representative isolate | Accession No. |
|---|---|---|---|---|---|---|---|
| G2a | 10/11/2021 | chicken | Akita | Northern | H5N8 | A/chicken/Akita/1T/2021 | EPI18720917 |
| G2a | 15/11/2021 | chicken | Kagoshima | Kyushu | H5N8 | A/chicken/Kagoshima/B1C/2021 | EPI18720919 |
| G2b | 13/11/2021 | chicken | Kagoshima | Kyushu | H5N1 | A/chicken/Kagoshima/21A1T/2021 | EPI18720695 |
| G2b | 17/11/2021 | chicken | Hyogo | Western | H5N1 | A/chicken/Hyogo/1T/2021 | EPI18720691 |
| G2b | 3/12/2021 | chicken | Kumamoto | Kyushu | H5N1 | A/chicken/Kumamoto/21A4C/2021 | EPI18720727 |
| G2b | 5/12/2021 | chicken | Chiba | Central | H5N1 | A/chicken/Chiba/21A1T/2021 | EPI18720718 |
| G2b | 7/12/2021 | chicken | Saitama | Central | H5N1 | A/chicken/Saitama/21A2T/2021 | EPI18720733 |
| G2b | 7/12/2021 | chicken | Hiroshima | Western | H5N1 | A/chicken/Hiroshima/21A3T/2021 | EPI18720753 |
| G2b | 12/12/2021 | chicken | Aomori | Northern | H5N1 | A/chicken/Aomori/21A1T/2021 | EPI18720743 |
| G2b | 31/12/2021 | chicken | Ehime | Shikoku | H5N1 | A/chicken/Ehime/21A2C/2021 | EPI18720767 |
| G2b | 4/1/2022 | chicken | Ehime | Shikoku | H5N1 | A/chicken/Ehime/21B2T/2022 | EPI18720769 |
| G2b | 4/1/2022 | chicken | Ehime | Shikoku | H5N1 | A/chicken/Ehime/21C5T/2022 | EPI18720778 |
| G2b | 13/1/2022 | chicken | Kagoshima | Kyushu | H5N1 | A/chicken/Kagoshima/21C2T/2022 | EPI18720780 |
| G2b | 19/1/2022 | chicken | Chiba | Central | H5N1 | A/chicken/Chiba/21B2T/2022 | EPI18720761 |
| G2b | 26/1/2022 | duck | Chiba | Central | H5N1 | A/duck/Chiba/21C1T/2022 | EPI18720794 |
| G2b | 13/5/2022 | chicken | Hokkaido | Hokkaido | H5N1 | A/chicken/Hokkaido/21D1T/2022 | EPI18720843 |
| G2d | 11/2/2022 | chicken | Iwate | Northern | H5N1 | A/chicken/Iwate/21A1T/2022 | EPI18720791 |
| G2d | 24/3/2022 | chicken | Miyagi | Northern | H5N1 | A/chicken/Miyagi/21A11T/2022 | EPI18720811 |
| G2d | 7/4/2022 | chicken | Aomori | Northern | H5N1 | A/chicken/Aomori/21B3T/2022 | EPI18720807 |
| G2d | 14/4/2022 | chicken | Aomori | Northern | H5N1 | A/chicken/Aomori/21C14T/2022 | EPI18720816 |
| G2d | 15/4/2022 | chicken | Hokkaido | Hokkaido | H5N1 | A/chicken/Hokkaido/21A4T/2022 | EPI18720826 |
| G2d | 16/4/2022 | emu | Hokkaido | Hokkaido | H5N1 | A/emu/Hokkaido/21B11T/2022 | EPI18720830 |
| G2d | 18/4/2022 | chicken | Akita | Northern | H5N1 | A/chicken/Akita/21B1T/2022 | EPI18720835 |
| G2d | 25/4/2022 | emu | Hokkaido | Hokkaido | H5N1 | A/emu/Hokkaido/21C1T/2022 | EPI18720866 |
| G2d | 11/5/2022 | emu | Iwate | Northern | H5N1 | A/emu/Iwate/21B1T/2022 | EPI18720867 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mine, J.; Takadate, Y.; Kumagai, A.; Sakuma, S.; Tsunekuni, R.; Miyazawa, K.; Uchida, Y. Genetics of H5N1 and H5N8 High-Pathogenicity Avian Influenza Viruses Isolated in Japan in Winter 2021–2022. Viruses 2024, 16, 358. https://doi.org/10.3390/v16030358
Mine J, Takadate Y, Kumagai A, Sakuma S, Tsunekuni R, Miyazawa K, Uchida Y. Genetics of H5N1 and H5N8 High-Pathogenicity Avian Influenza Viruses Isolated in Japan in Winter 2021–2022. Viruses. 2024; 16(3):358. https://doi.org/10.3390/v16030358
Chicago/Turabian StyleMine, Junki, Yoshihiro Takadate, Asuka Kumagai, Saki Sakuma, Ryota Tsunekuni, Kohtaro Miyazawa, and Yuko Uchida. 2024. "Genetics of H5N1 and H5N8 High-Pathogenicity Avian Influenza Viruses Isolated in Japan in Winter 2021–2022" Viruses 16, no. 3: 358. https://doi.org/10.3390/v16030358
APA StyleMine, J., Takadate, Y., Kumagai, A., Sakuma, S., Tsunekuni, R., Miyazawa, K., & Uchida, Y. (2024). Genetics of H5N1 and H5N8 High-Pathogenicity Avian Influenza Viruses Isolated in Japan in Winter 2021–2022. Viruses, 16(3), 358. https://doi.org/10.3390/v16030358