
IFNα Subtypes in HIV Infection and Immunity

 ,
,
Abstract
1. Introduction
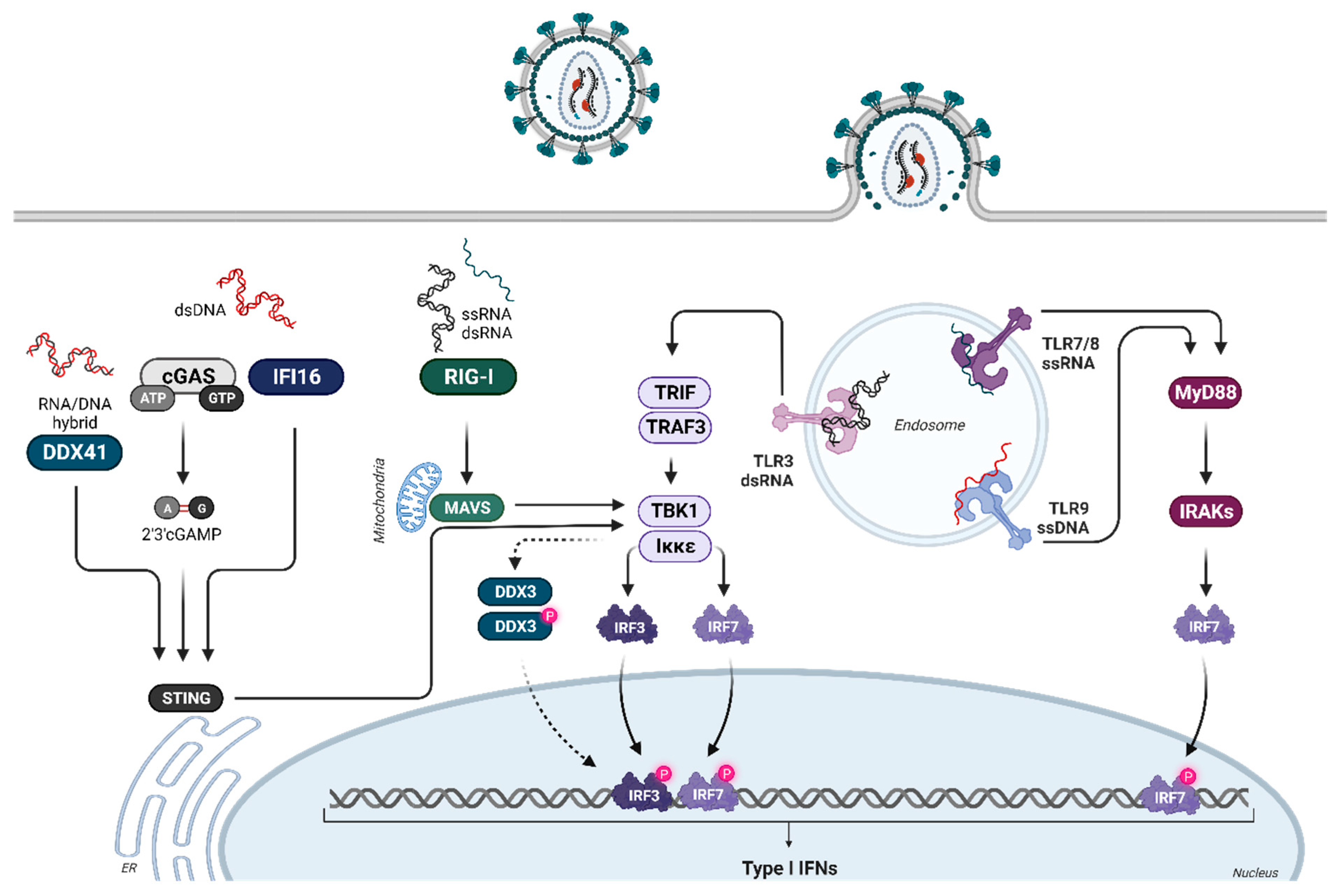
2. Induction of IFNα Subtypes during Retroviral Infections
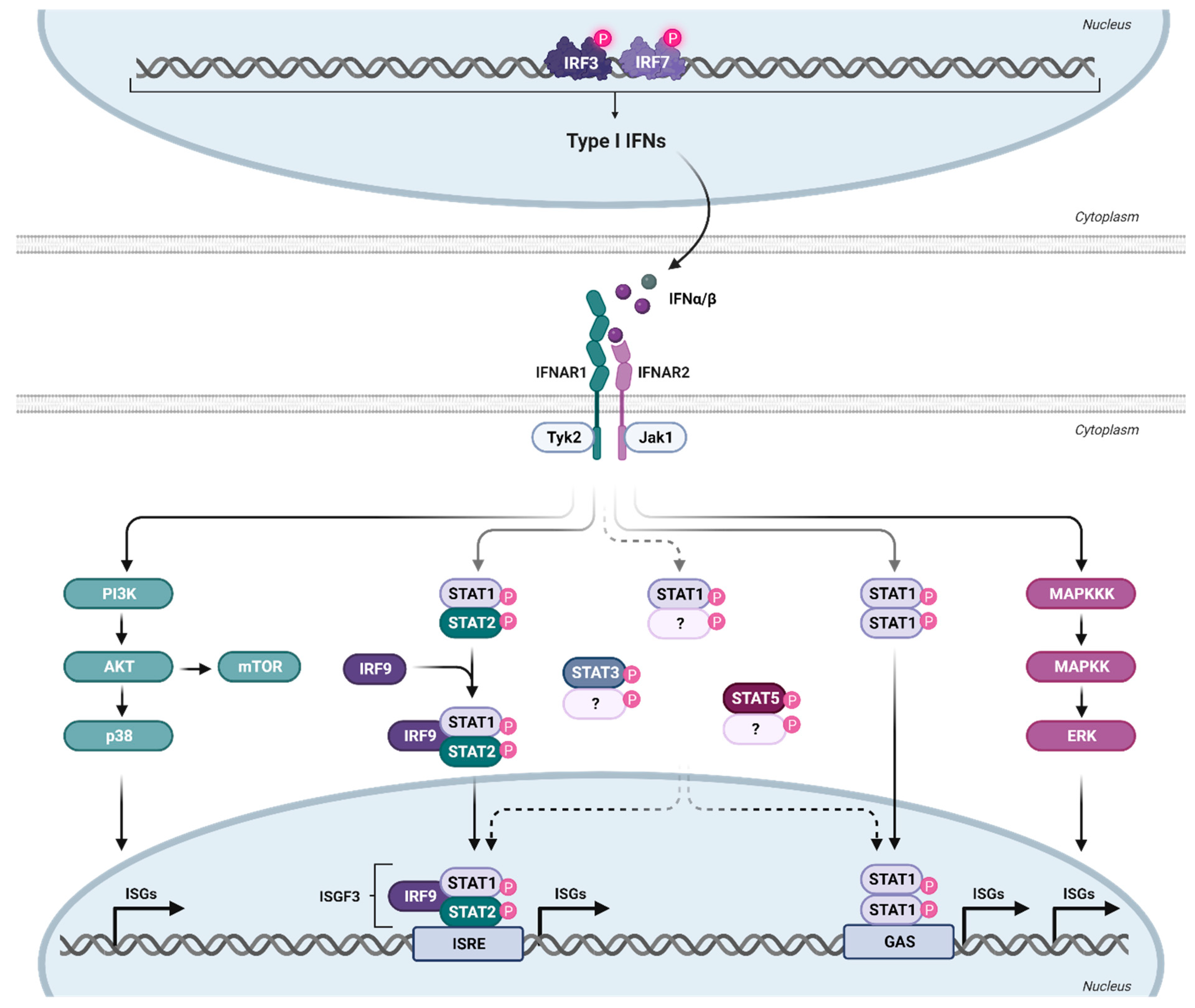
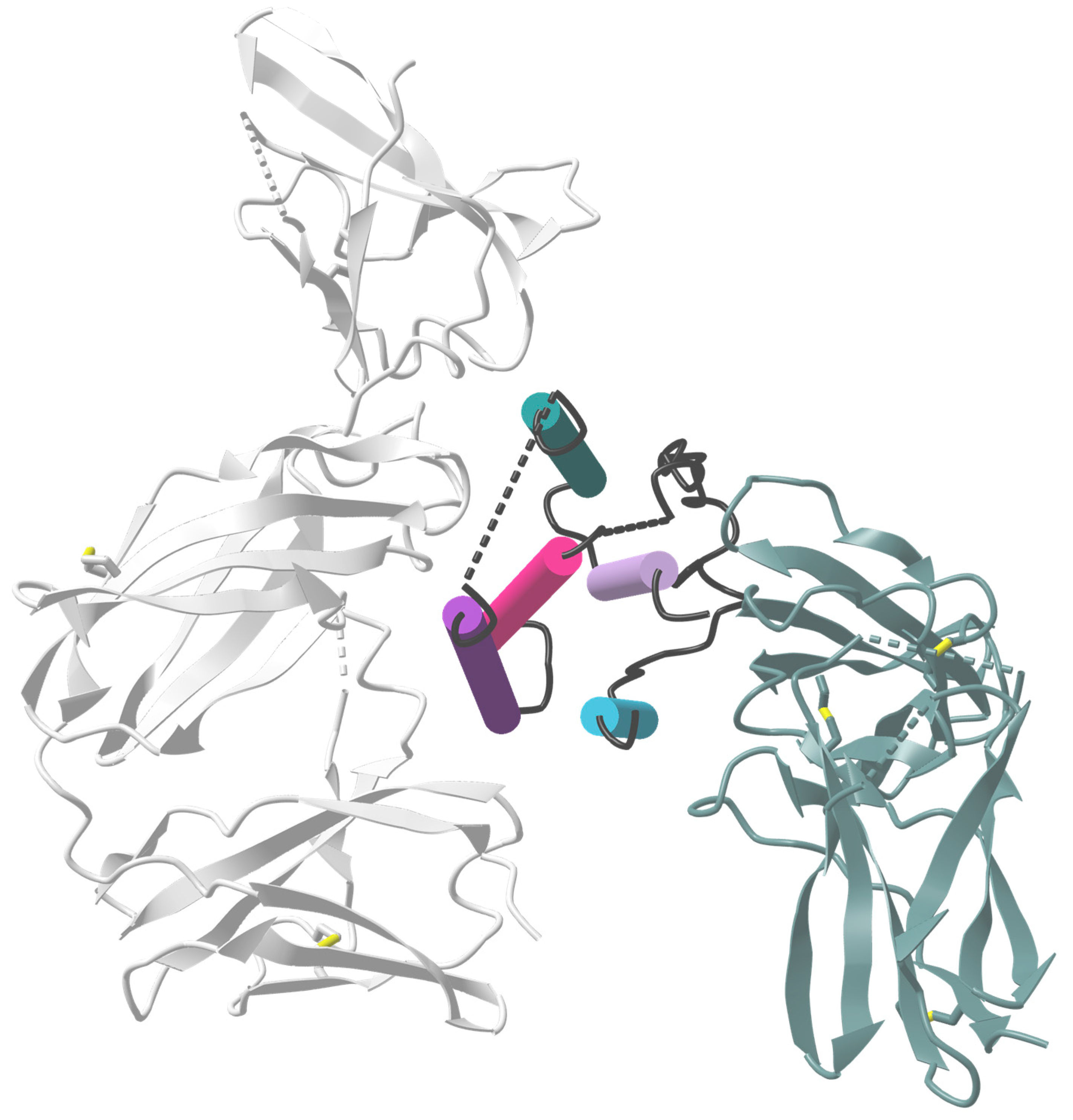
3. IFNα Subtype-Mediated Downstream Signaling and ISG Expression Pattern during HIV Infection
4. Antiviral Activity of IFNα Subtypes during Retroviral Infections
5. Modulation of Immune Cell Functions by IFNα Subtypes during Retroviral Infections
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Trilling, M.; Bellora, N.; Rutkowski, A.J.; de Graaf, M.; Dickinson, P.; Robertson, K.; da Costa, O.P.; Ghazal, P.; Friedel, C.C.; Alba, M.M.; et al. Deciphering the modulation of gene expression by type I and II interferons combining 4sU-tagging, translational arrest and in silico promoter analysis. Nucleic Acids Res. 2013, 41, 8107–8125. [Google Scholar] [CrossRef]
- Megger, D.A.; Philipp, J.; Le-Trilling, V.T.K.; Sitek, B.; Trilling, M. Deciphering of the Human Interferon-Regulated Proteome by Mass Spectrometry-Based Quantitative Analysis Reveals Extent and Dynamics of Protein Induction and Repression. Front. Immunol. 2017, 8, 1139. [Google Scholar] [CrossRef] [PubMed]
- Sertznig, H.; Roesmann, F.; Wilhelm, A.; Heininger, D.; Bleekmann, B.; Elsner, C.; Santiago, M.; Schuhenn, J.; Karakoese, Z.; Benatzy, Y.; et al. SRSF1 acts as an IFN-I-regulated cellular dependency factor decisively affecting HIV-1 post-integration steps. Front. Immunol. 2022, 13, 935800. [Google Scholar] [CrossRef] [PubMed]
- van Pesch, V.; Lanaya, H.; Renauld, J.C.; Michiels, T. Characterization of the murine alpha interferon gene family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef] [PubMed]
- Woelk, C.H.; Frost, S.D.; Richman, D.D.; Higley, P.E.; Kosakovsky Pond, S.L. Evolution of the interferon alpha gene family in eutherian mammals. Gene 2007, 397, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, L.; Liu, W. Distinct evolution process among type I interferon in mammals. Protein Cell 2013, 4, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.O.; Pomykala, H.M.; Bohlander, S.K.; Maltepe, E.; Malik, K.; Brownstein, B.; Olopade, O.I. Structure of the human type-I interferon gene cluster determined from a YAC clone contig. Genomics 1994, 22, 540–552. [Google Scholar] [CrossRef]
- Genin, P.; Lin, R.; Hiscott, J.; Civas, A. Differential regulation of human interferon A gene expression by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 2009, 29, 3435–3450. [Google Scholar] [CrossRef] [PubMed]
- Freaney, J.E.; Zhang, Q.; Yigit, E.; Kim, R.; Widom, J.; Wang, J.P.; Horvath, C.M. High-density nucleosome occupancy map of human chromosome 9p21-22 reveals chromatin organization of the type I interferon gene cluster. J. Interferon Cytokine Res. 2014, 34, 676–685. [Google Scholar] [CrossRef]
- Hardy, M.P.; Owczarek, C.M.; Jermiin, L.S.; Ejdebäck, M.; Hertzog, P.J. Characterization of the type I interferon locus and identification of novel genes. Genomics 2004, 84, 331–345. [Google Scholar] [CrossRef]
- Zwarthoff, E.C.; Mooren, A.T.; Trapman, J. Organization, structure and expression of murine interferon alpha genes. Nucleic Acids Res. 1985, 13, 791–804. [Google Scholar] [CrossRef]
- Lavoie, T.B.; Kalie, E.; Crisafulli-Cabatu, S.; Abramovich, R.; DiGioia, G.; Moolchan, K.; Pestka, S.; Schreiber, G. Binding and activity of all human alpha interferon subtypes. Cytokine 2011, 56, 282–289. [Google Scholar] [CrossRef]
- Cull, V.S.; Tilbrook, P.A.; Bartlett, E.J.; Brekalo, N.L.; James, C.M. Type I interferon differential therapy for erythroleukemia: Specificity of STAT activation. Blood 2003, 101, 2727–2735. [Google Scholar] [CrossRef]
- Karakoese, Z.; Le-Trilling, V.T.; Schuhenn, J.; Francois, S.; Lu, M.; Liu, J.; Trilling, M.; Hoffmann, D.; Dittmer, U.; Sutter, K. Targeted mutations in IFNalpha2 improve its antiviral activity against various viruses. mBio 2023, 14, e02357-23. [Google Scholar] [CrossRef]
- Tomasello, E.; Pollet, E.; Vu Manh, T.P.; Uze, G.; Dalod, M. Harnessing Mechanistic Knowledge on Beneficial Versus Deleterious IFN-I Effects to Design Innovative Immunotherapies Targeting Cytokine Activity to Specific Cell Types. Front. Immunol. 2014, 5, 526. [Google Scholar] [CrossRef]
- Moll, H.P.; Maier, T.; Zommer, A.; Lavoie, T.; Brostjan, C. The differential activity of interferon-alpha subtypes is consistent among distinct target genes and cell types. Cytokine 2011, 53, 52–59. [Google Scholar] [CrossRef]
- Gibbert, K.; Joedicke, J.J.; Meryk, A.; Trilling, M.; Francois, S.; Duppach, J.; Kraft, A.; Lang, K.S.; Dittmer, U. Interferon-alpha subtype 11 activates NK cells and enables control of retroviral infection. PLoS Pathog. 2012, 8, e1002868. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, N.; Gibbert, K.; Alter, C.; Nair, S.; Zelinskyy, G.; James, C.M.; Dittmer, U. Anti-retroviral effects of type I IFN subtypes in vivo. Eur. J. Immunol. 2009, 39, 136–146. [Google Scholar] [CrossRef]
- Scagnolari, C.; Trombetti, S.; Selvaggi, C.; Carbone, T.; Monteleone, K.; Spano, L.; Di Marco, P.; Pierangeli, A.; Maggi, F.; Riva, E.; et al. In vitro sensitivity of human metapneumovirus to type I interferons. Viral Immunol. 2011, 24, 159–164. [Google Scholar] [CrossRef]
- Cull, V.S.; Bartlett, E.J.; James, C.M. Type I interferon gene therapy protects against cytomegalovirus-induced myocarditis. Immunology 2002, 106, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Härle, P.; Cull, V.; Agbaga, M.P.; Silverman, R.; Williams, B.R.; James, C.; Carr, D.J. Differential effect of murine alpha/beta interferon transgenes on antagonization of herpes simplex virus type 1 replication. J. Virol. 2002, 76, 6558–6567. [Google Scholar] [CrossRef]
- Song, J.; Li, S.; Zhou, Y.; Liu, J.; Francois, S.; Lu, M.; Yang, D.; Dittmer, U.; Sutter, K. Different antiviral effects of IFNα subtypes in a mouse model of HBV infection. Sci. Rep. 2017, 7, 334. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Lai, F.; Wang, Y.; Sutter, K.; Dittmer, U.; Ye, J.; Zai, W.; Liu, M.; Shen, F.; et al. Functional Comparison of Interferon-α Subtypes Reveals Potent Hepatitis B Virus Suppression by a Concerted Action of Interferon-α and Interferon-γ Signaling. Hepatology 2021, 73, 486–502. [Google Scholar] [CrossRef]
- Schmitz, Y.; Schwerdtfeger, M.; Westmeier, J.; Littwitz-Salomon, E.; Alt, M.; Brochhagen, L.; Krawczyk, A.; Sutter, K. Superior antiviral activity of IFNbeta in genital HSV-1 infection. Front. Cell. Infect. Microbiol. 2022, 12, 949036. [Google Scholar] [CrossRef]
- Schuhenn, J.; Meister, T.L.; Todt, D.; Bracht, T.; Schork, K.; Billaud, J.N.; Elsner, C.; Heinen, N.; Karakoese, Z.; Haid, S.; et al. Differential interferon-alpha subtype induced immune signatures are associated with suppression of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2022, 119, e2111600119. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar] [CrossRef]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef]
- Genin, P.; Vaccaro, A.; Civas, A. The role of differential expression of human interferon—A genes in antiviral immunity. Cytokine Growth Factor. Rev. 2009, 20, 283–295. [Google Scholar] [CrossRef]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Taubert, D.; Jung, N.; Fatkenheuer, G.; van Lunzen, J.; Hartmann, P.; Romerio, F. Preferential upregulation of interferon-alpha subtype 2 expression in HIV-1 patients. AIDS Res. Hum. Retroviruses 2009, 25, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, B.; Esser, S.; Jessen, H.; Streeck, H.; Widera, M.; Yang, R.; Dittmer, U.; Sutter, K. Expression Pattern of Individual IFNA Subtypes in Chronic HIV Infection. J. Interferon Cytokine Res. 2017, 37, 541–549. [Google Scholar] [CrossRef]
- Harper, M.S.; Guo, K.; Gibbert, K.; Lee, E.J.; Dillon, S.M.; Barrett, B.S.; McCarter, M.D.; Hasenkrug, K.J.; Dittmer, U.; Wilson, C.C.; et al. Interferon-α Subtypes in an Ex Vivo Model of Acute HIV-1 Infection: Expression, Potency and Effector Mechanisms. PLoS Pathog. 2015, 11, e1005254. [Google Scholar] [CrossRef]
- Dillon, S.M.; Guo, K.; Austin, G.L.; Gianella, S.; Engen, P.A.; Mutlu, E.A.; Losurdo, J.; Swanson, G.; Chakradeo, P.; Keshavarzian, A.; et al. A compartmentalized type I interferon response in the gut during chronic HIV-1 infection is associated with immunopathogenesis. AIDS 2018, 32, 1599–1611. [Google Scholar] [CrossRef]
- Dutrieux, J.; Fabre-Mersseman, V.; Charmeteau-De Muylder, B.; Rancez, M.; Ponte, R.; Rozlan, S.; Figueiredo-Morgado, S.; Bernard, A.; Beq, S.; Couedel-Courteille, A.; et al. Modified interferon-alpha subtypes production and chemokine networks in the thymus during acute simian immunodeficiency virus infection, impact on thymopoiesis. AIDS 2014, 28, 1101–1113. [Google Scholar] [CrossRef]
- Easlick, J.; Szubin, R.; Lantz, S.; Baumgarth, N.; Abel, K. The early interferon alpha subtype response in infant macaques infected orally with SIV. J. Acquir. Immune Defic. Syndr. 2010, 55, 14–28. [Google Scholar] [CrossRef]
- Zaritsky, L.A.; Dery, A.; Leong, W.Y.; Gama, L.; Clements, J.E. Tissue-specific interferon alpha subtype response to SIV infection in brain, spleen, and lung. J. Interferon Cytokine Res. 2013, 33, 24–33. [Google Scholar] [CrossRef]
- Rodero, M.P.; Decalf, J.; Bondet, V.; Hunt, D.; Rice, G.I.; Werneke, S.; McGlasson, S.L.; Alyanakian, M.A.; Bader-Meunier, B.; Barnerias, C.; et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med. 2017, 214, 1547–1555. [Google Scholar] [CrossRef]
- Jaks, E.; Gavutis, M.; Uze, G.; Martal, J.; Piehler, J. Differential receptor subunit affinities of type I interferons govern differential signal activation. J. Mol. Biol. 2007, 366, 525–539. [Google Scholar] [CrossRef]
- Piehler, J.; Thomas, C.; Garcia, K.C.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Vivian, J.P.; Nguyen, T.K.; Mangan, N.E.; Gould, J.A.; Braniff, S.J.; Zaker-Tabrizi, L.; Fung, K.Y.; Forster, S.C.; Beddoe, T.; et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat. Immunol. 2013, 14, 901–907. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and interferons: Who’s interfering with whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef]
- Suprunenko, T.; Hofer, M.J. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Factor. Rev. 2016, 29, 35–43. [Google Scholar] [CrossRef]
- Leviyang, S. Interferon stimulated binding of ISRE is cell type specific and is predicted by homeostatic chromatin state. Cytokine X 2021, 3, 100056. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef]
- Lavender, K.J.; Gibbert, K.; Peterson, K.E.; Van Dis, E.; Francois, S.; Woods, T.; Messer, R.J.; Gawanbacht, A.; Müller, J.A.; Münch, J.; et al. Interferon Alpha Subtype-Specific Suppression of HIV-1 Infection In Vivo. J. Virol. 2016, 90, 6001–6013. [Google Scholar] [CrossRef]
- Guo, K.; Shen, G.; Kibbie, J.; Gonzalez, T.; Dillon, S.M.; Smith, H.A.; Cooper, E.H.; Lavender, K.; Hasenkrug, K.J.; Sutter, K.; et al. Qualitative Differences Between the IFNα subtypes and IFNβ Influence Chronic Mucosal HIV-1 Pathogenesis. PLoS Pathog. 2020, 16, e1008986. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. The p38 mitogen-activated protein kinase pathway and its role in interferon signaling. Pharmacol. Ther. 2003, 98, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef]
- Zhao, L.J.; Wang, W.; Wang, W.B.; Ren, H.; Qi, Z.T. Involvement of ERK pathway in interferon alpha-mediated antiviral activity against hepatitis C virus. Cytokine 2015, 72, 17–24. [Google Scholar] [CrossRef]
- Furler, R.L.; Uittenbogaart, C.H. Signaling through the P38 and ERK pathways: A common link between HIV replication and the immune response. Immunol. Res. 2010, 48, 99–109. [Google Scholar] [CrossRef]
- Van der Sluis, R.M.; Zerbato, J.M.; Rhodes, J.W.; Pascoe, R.D.; Solomon, A.; Kumar, N.A.; Dantanarayana, A.I.; Tennakoon, S.; Dufloo, J.; McMahon, J.; et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PLoS Pathog. 2020, 16, e1008151. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Urban, J. A review on recent trends in the phosphoproteomics workflow. From sample preparation to data analysis. Anal. Chim. Acta 2022, 1199, 338857. [Google Scholar] [CrossRef]
- Wojcechowskyj, J.A.; Didigu, C.A.; Lee, J.Y.; Parrish, N.F.; Sinha, R.; Hahn, B.H.; Bushman, F.D.; Jensen, S.T.; Seeholzer, S.H.; Doms, R.W. Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe 2013, 13, 613–623. [Google Scholar] [CrossRef]
- Karakoese, Z.; Schwerdtfeger, M.; Karsten, C.B.; Esser, S.; Dittmer, U.; Sutter, K. Distinct Type I Interferon Subtypes Differentially Stimulate T Cell Responses in HIV-1-Infected Individuals. Front. Immunol. 2022, 13, 936918. [Google Scholar] [CrossRef]
- Dickow, J.; Francois, S.; Kaiserling, R.L.; Malyshkina, A.; Drexler, I.; Westendorf, A.M.; Lang, K.S.; Santiago, M.L.; Dittmer, U.; Sutter, K. Diverse Immunomodulatory Effects of Individual IFNα Subtypes on Virus-Specific CD8+ T Cell Responses. Front. Immunol. 2019, 10, 2255. [Google Scholar] [CrossRef]
- Sperber, S.J.; Gocke, D.J.; Haberzettl, C.; Kuk, R.; Schwartz, B.; Pestka, S. Anti-HIV-1 activity of recombinant and hybrid species of interferon-alpha. J. Interferon Res. 1992, 12, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Katabira, E.T.; Sewankambo, N.K.; Mugerwa, R.D.; Belsey, E.M.; Mubiru, F.X.; Othieno, C.; Kataaha, P.; Karam, M.; Youle, M.; Perriens, J.H.; et al. Lack of efficacy of low dose oral interferon alfa in symptomatic HIV-1 infection: A randomised, double blind, placebo controlled trial. Sex. Transm. Infect. 1998, 74, 265–270. [Google Scholar] [CrossRef]
- Alston, B.; Ellenberg, J.H.; Standiford, H.C.; Muth, K.; Martinez, A.; Greaves, W.; Kumi, J. A multicenter, randomized, controlled trial of three preparations of low-dose oral alpha-interferon in HIV-infected patients with CD4+ counts between 50 and 350 cells/mm(3). Division of AIDS Treatment Research Initiative (DATRI) 022 Study Group. J. Acquir. Immune Defic. Syndr. 1999, 22, 348–357. [Google Scholar] [CrossRef]
- Azzoni, L.; Foulkes, A.S.; Papasavvas, E.; Mexas, A.M.; Lynn, K.M.; Mounzer, K.; Tebas, P.; Jacobson, J.M.; Frank, I.; Busch, M.P.; et al. Pegylated Interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J. Infect. Dis. 2013, 207, 213–222. [Google Scholar] [CrossRef]
- Asmuth, D.M.; Murphy, R.L.; Rosenkranz, S.L.; Lertora, J.J.; Kottilil, S.; Cramer, Y.; Chan, E.S.; Schooley, R.T.; Rinaldo, C.R.; Thielman, N.; et al. Safety, tolerability, and mechanisms of antiretroviral activity of pegylated interferon Alfa-2a in HIV-1-monoinfected participants: A phase II clinical trial. J. Infect. Dis. 2010, 201, 1686–1696. [Google Scholar] [CrossRef]
- Tauzin, A.; Espinosa Ortiz, A.; Blake, O.; Soundaramourty, C.; Joly-Beauparlant, C.; Nicolas, A.; Droit, A.; Dutrieux, J.; Estaquier, J.; Mammano, F. Differential Inhibition of HIV Replication by the 12 Interferon Alpha Subtypes. J. Virol. 2021, 95, e0231120. [Google Scholar] [CrossRef]
- Vazquez, N.; Schmeisser, H.; Dolan, M.A.; Bekisz, J.; Zoon, K.C.; Wahl, S.M. Structural variants of IFNalpha preferentially promote antiviral functions. Blood 2011, 118, 2567–2577. [Google Scholar] [CrossRef]
- Kalie, E.; Jaitin, D.A.; Abramovich, R.; Schreiber, G. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J. Biol. Chem. 2007, 282, 11602–11611. [Google Scholar] [CrossRef]
- Thomas, C.; Moraga, I.; Levin, D.; Krutzik, P.O.; Podoplelova, Y.; Trejo, A.; Lee, C.; Yarden, G.; Vleck, S.E.; Glenn, J.S.; et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 2011, 146, 621–632. [Google Scholar] [CrossRef]
- Schwerdtfeger, M.; Dickow, J.; Schmitz, Y.; Francois, S.; Karakoese, Z.; Malyshkina, A.; Knuschke, T.; Dittmer, U.; Sutter, K. Immunotherapy With Interferon alpha11, But Not Interferon Beta, Controls Persistent Retroviral Infection. Front. Immunol. 2021, 12, 809774. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Choi, J.G.; Ortega, N.M.; Zhang, J.; Shankar, P.; Swamy, N.M. Gene therapy with plasmids encoding IFN-beta or IFN-alpha14 confers long-term resistance to HIV-1 in humanized mice. Oncotarget 2016, 7, 78412–78420. [Google Scholar] [CrossRef] [PubMed]
- Sutter, K.; Lavender, K.J.; Messer, R.J.; Widera, M.; Williams, K.; Race, B.; Hasenkrug, K.J.; Dittmer, U. Concurrent administration of IFNalpha14 and cART in TKO-BLT mice enhances suppression of HIV-1 viremia but does not eliminate the latent reservoir. Sci. Rep. 2019, 9, 18089. [Google Scholar] [CrossRef] [PubMed]
- Gruenbach, M.; Muller, C.K.S.; Schlaepfer, E.; Baroncini, L.; Russenberger, D.; Kadzioch, N.P.; Escher, B.; Schlapschy, M.; Skerra, A.; Bredl, S.; et al. cART Restores Transient Responsiveness to IFN Type 1 in HIV-Infected Humanized Mice. J. Virol. 2022, 96, e0082722. [Google Scholar] [CrossRef]
- Sandler, N.G.; Bosinger, S.E.; Estes, J.D.; Zhu, R.T.; Tharp, G.K.; Boritz, E.; Levin, D.; Wijeyesinghe, S.; Makamdop, K.N.; del Prete, G.Q.; et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 2014, 511, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Carnathan, D.; Lawson, B.; Yu, J.; Patel, K.; Billingsley, J.M.; Tharp, G.K.; Delmas, O.M.; Dawoud, R.; Wilkinson, P.; Nicolette, C.; et al. Reduced Chronic Lymphocyte Activation following Interferon Alpha Blockade during the Acute Phase of Simian Immunodeficiency Virus Infection in Rhesus Macaques. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Vanderford, T.H.; Slichter, C.; Rogers, K.A.; Lawson, B.O.; Obaede, R.; Else, J.; Villinger, F.; Bosinger, S.E.; Silvestri, G. Treatment of SIV-infected sooty mangabeys with a type-I IFN agonist results in decreased virus replication without inducing hyperimmune activation. Blood 2012, 119, 5750–5757. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Ma, J.; Li, J.; Li, D.; Li, G.; Li, F.; Zhang, Q.; Yu, H.; Yasui, F.; Ye, C.; et al. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J. Clin. Investig. 2017, 127, 269–279. [Google Scholar] [CrossRef]
- Zhen, A.; Rezek, V.; Youn, C.; Lam, B.; Chang, N.; Rick, J.; Carrillo, M.; Martin, H.; Kasparian, S.; Syed, P.; et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Investig. 2017, 127, 260–268. [Google Scholar] [CrossRef]
- Nganou-Makamdop, K.; Billingsley, J.M.; Yaffe, Z.; O’Connor, G.; Tharp, G.K.; Ransier, A.; Laboune, F.; Matus-Nicodemos, R.; Lerner, A.; Gharu, L.; et al. Type I IFN signaling blockade by a PASylated antagonist during chronic SIV infection suppresses specific inflammatory pathways but does not alter T cell activation or virus replication. PLoS Pathog. 2018, 14, e1007246. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Ng, C.T.; Sullivan, B.M.; Teijaro, J.R.; Lee, A.M.; Welch, M.; Rice, S.; Sheehan, K.C.; Schreiber, R.D.; Oldstone, M.B. Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 2015, 17, 653–661. [Google Scholar] [CrossRef]
- Swainson, L.A.; Sharma, A.A.; Ghneim, K.; Ribeiro, S.P.; Wilkinson, P.; Dunham, R.M.; Albright, R.G.; Wong, S.; Estes, J.D.; Piatak, M.; et al. IFN-alpha blockade during ART-treated SIV infection lowers tissue vDNA, rescues immune function, and improves overall health. JCI Insight 2022, 7, e153046. [Google Scholar] [CrossRef]
- Wang, B.X.; Fish, E.N. The yin and yang of viruses and interferons. Trends Immunol. 2012, 33, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef]
- Kuka, M.; De Giovanni, M.; Iannacone, M. The role of type I interferons in CD4+ T cell differentiation. Immunol. Lett. 2019, 215, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Cha, L.; Berry, C.M.; Nolan, D.; Castley, A.; Fernandez, S.; French, M.A. Interferon-alpha, immune activation and immune dysfunction in treated HIV infection. Clin. Transl. Immunol. 2014, 3, e10. [Google Scholar] [CrossRef]
- Jacquelin, B.; Petitjean, G.; Kunkel, D.; Liovat, A.S.; Jochems, S.P.; Rogers, K.A.; Ploquin, M.J.; Madec, Y.; Barre-Sinoussi, F.; Dereuddre-Bosquet, N.; et al. Innate immune responses and rapid control of inflammation in African green monkeys treated or not with interferon-alpha during primary SIVagm infection. PLoS Pathog. 2014, 10, e1004241. [Google Scholar] [CrossRef]
- Herbeuval, J.P.; Boasso, A.; Grivel, J.C.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Chougnet, C.; Lifson, J.D.; Shearer, G.M. TNF-related apoptosis-inducing ligand (TRAIL) in HIV-1-infected patients and its in vitro production by antigen-presenting cells. Blood 2005, 105, 2458–2464. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Mueller, Y.M.; Yang, G.; Boesteanu, A.C.; Gracias, D.T.; Do, D.H.; Hope, J.L.; Kathuria, N.; McGettigan, S.E.; Lewis, M.G.; et al. Type I interferon upregulates Bak and contributes to T cell loss during human immunodeficiency virus (HIV) infection. PLoS Pathog. 2013, 9, e1003658. [Google Scholar] [CrossRef]
- Fernandez, S.; Tanaskovic, S.; Helbig, K.; Rajasuriar, R.; Kramski, M.; Murray, J.M.; Beard, M.; Purcell, D.; Lewin, S.R.; Price, P.; et al. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J. Infect. Dis. 2011, 204, 1927–1935. [Google Scholar] [CrossRef]
- Bosinger, S.E.; Utay, N.S. Type I interferon: Understanding its role in HIV pathogenesis and therapy. Curr. HIV/AIDS Rep. 2015, 12, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhang, T.; Wang, R.; Zhang, H.; Huang, X.; Yin, J.; Zhang, L.; Xu, X.; Wu, H. Plasma IP-10 is associated with rapid disease progression in early HIV-1 infection. Viral Immunol. 2012, 25, 333–337. [Google Scholar] [CrossRef]
- Rout, S.S.; Di, Y.; Dittmer, U.; Sutter, K.; Lavender, K.J. Distinct effects of treatment with two different interferon-alpha subtypes on HIV-1-associated T-cell activation and dysfunction in humanized mice. AIDS 2022, 36, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Lichtfuss, G.F.; Meehan, A.C.; Cheng, W.J.; Cameron, P.U.; Lewin, S.R.; Crowe, S.M.; Jaworowski, A. HIV inhibits early signal transduction events triggered by CD16 cross-linking on NK cells, which are important for antibody-dependent cellular cytotoxicity. J. Leukoc. Biol. 2011, 89, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Chang, J.J.; Dantanarayana, A.I.; Solomon, A.; Evans, V.A.; Pascoe, R.; Gubser, C.; Trautman, L.; Fromentin, R.; Chomont, N.; et al. Combination Immune Checkpoint Blockade Enhances IL-2 and CD107a Production from HIV-Specific T Cells Ex Vivo in People Living with HIV on Antiretroviral Therapy. J. Immunol. 2022, 208, 54–62. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Cell Type | HIV Infection/ Exposure | Upregulated IFNA mRNAs | Detection Method | References |
|---|---|---|---|---|
| Isolated pDCs | Patients with HIV (CDC stage A or C) | IFNA6 and IFNA2 | RT-PCR | [35] |
| Patients with HIV (CDC stage C) | IFNA1/13, IFNA8, IFNA14, IFNA16, IFNA17, and IFNA21 | |||
| PBMCs | ART-treated chronically HIV-positive patients | IFNA2, IFNA4, IFNA5, IFNA6, IFNA7, IFNA14, and IFNA16 | RT-PCR | [36] |
| ART-naïve chronically HIV-positive patients | ||||
| PBMCs | ART-naïve chronically HIV-positive patients | IFNA1/13, IFNA2, IFNA5, and IFNA4 | Illumina sequencing | [38] |
| Isolated pDCs | Exposed to HIV-1BaL | IFNA2 and IFNA14 | Illumina sequencing | [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakoese, Z.; Ingola, M.; Sitek, B.; Dittmer, U.; Sutter, K. IFNα Subtypes in HIV Infection and Immunity. Viruses 2024, 16, 364. https://doi.org/10.3390/v16030364
Karakoese Z, Ingola M, Sitek B, Dittmer U, Sutter K. IFNα Subtypes in HIV Infection and Immunity. Viruses. 2024; 16(3):364. https://doi.org/10.3390/v16030364
Chicago/Turabian StyleKarakoese, Zehra, Martha Ingola, Barbara Sitek, Ulf Dittmer, and Kathrin Sutter. 2024. "IFNα Subtypes in HIV Infection and Immunity" Viruses 16, no. 3: 364. https://doi.org/10.3390/v16030364
APA StyleKarakoese, Z., Ingola, M., Sitek, B., Dittmer, U., & Sutter, K. (2024). IFNα Subtypes in HIV Infection and Immunity. Viruses, 16(3), 364. https://doi.org/10.3390/v16030364