

CD8+ T Cells Mediate Lethal Lung Pathology in the Absence of PD-L1 and Type I Interferon Signalling following LCMV Infection

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. LCMV Infection
2.3. Monoclonal Antibody Neutralisation of CD8, Ly6G and PD-L1
2.4. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction
2.5. RNase Protection Assay
2.6. Histology of Liver and Lung Tissue
2.7. Quantification of Plasma Cytokines
2.8. Flow Cytometry
2.9. tSNE Analysis
2.10. Statistical Analysis
3. Results
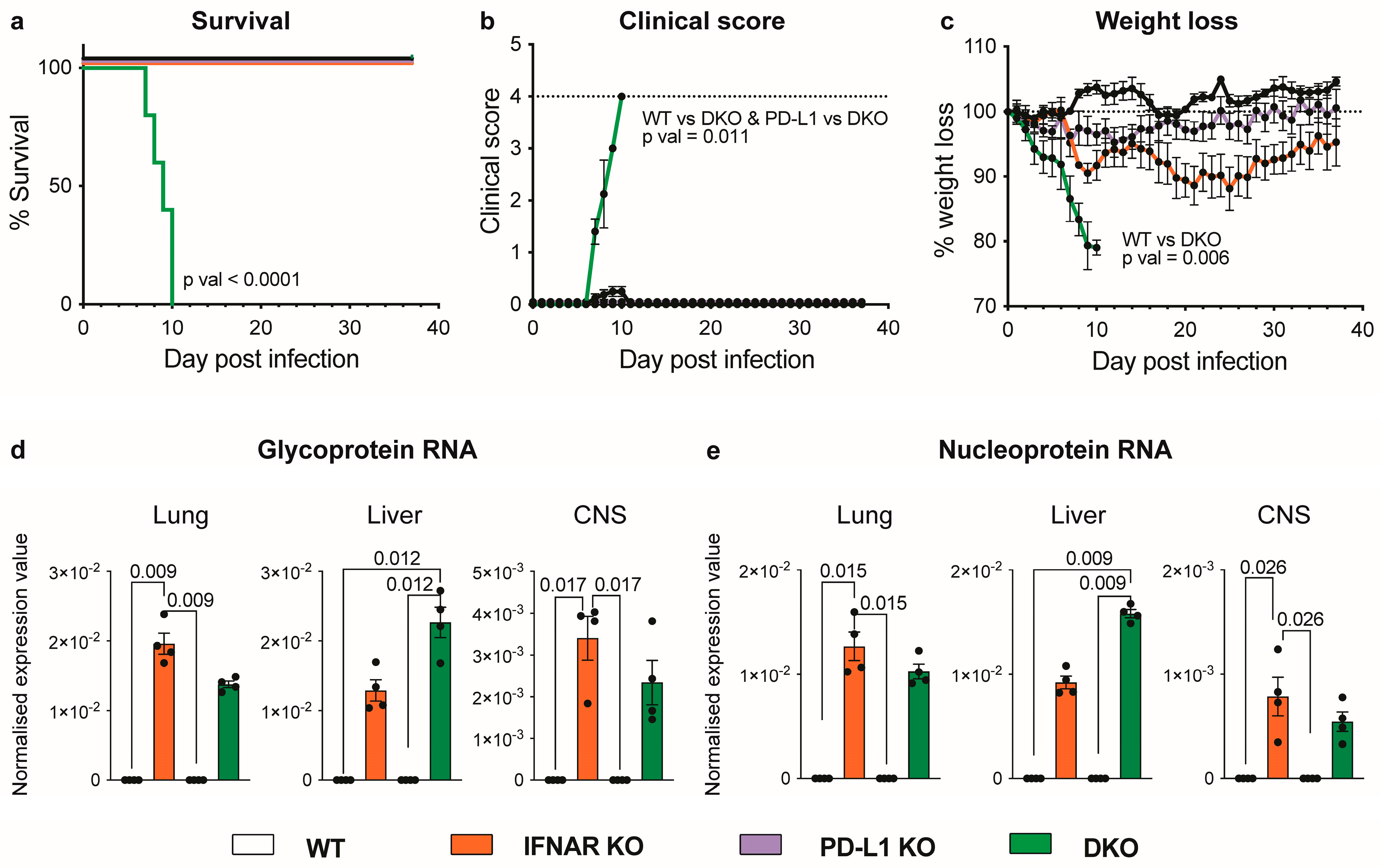
3.1. Absence of IFNAR1 and PD-L1 Signalling Results in Lethal Disease following LCMV Infection
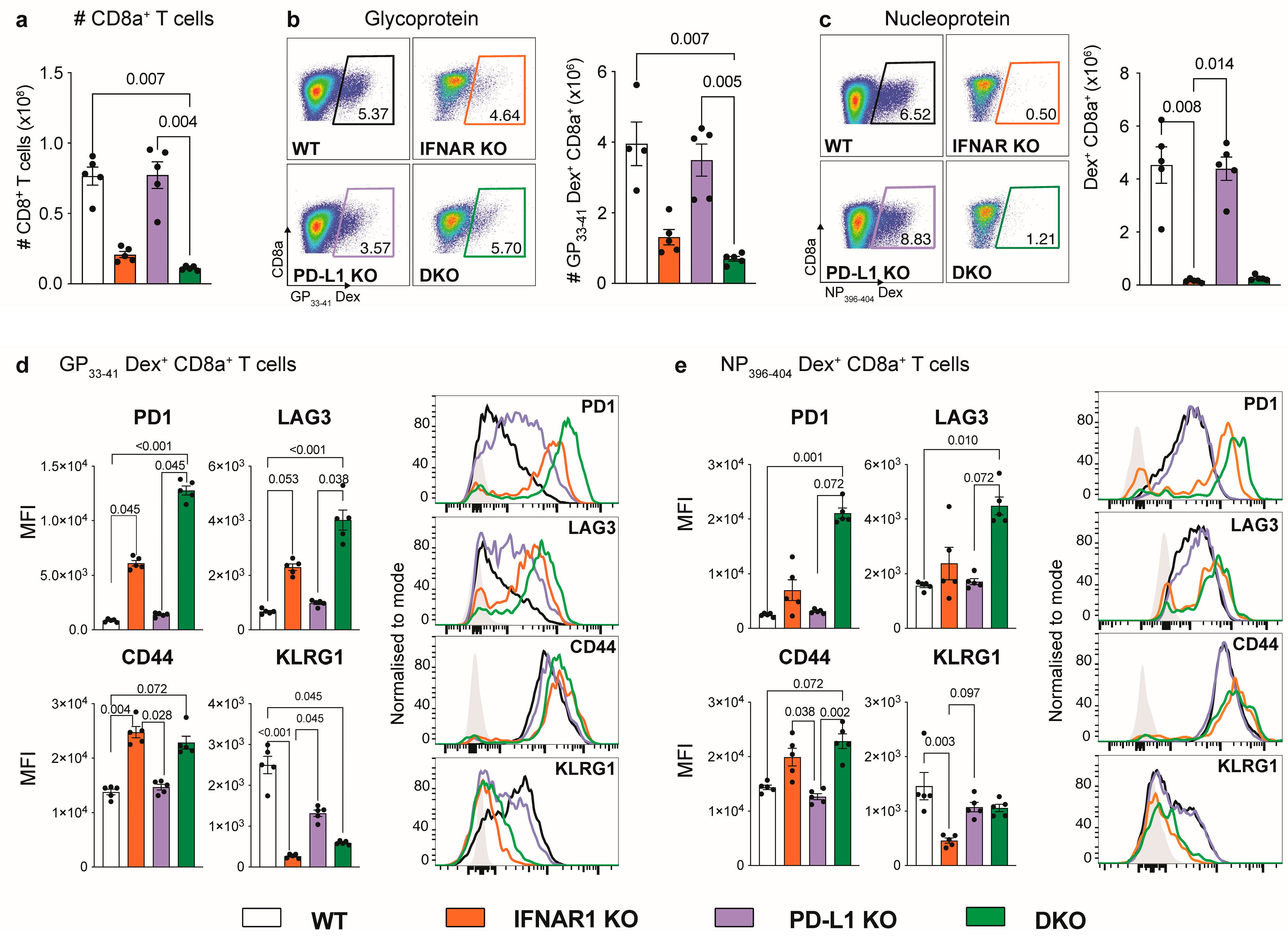
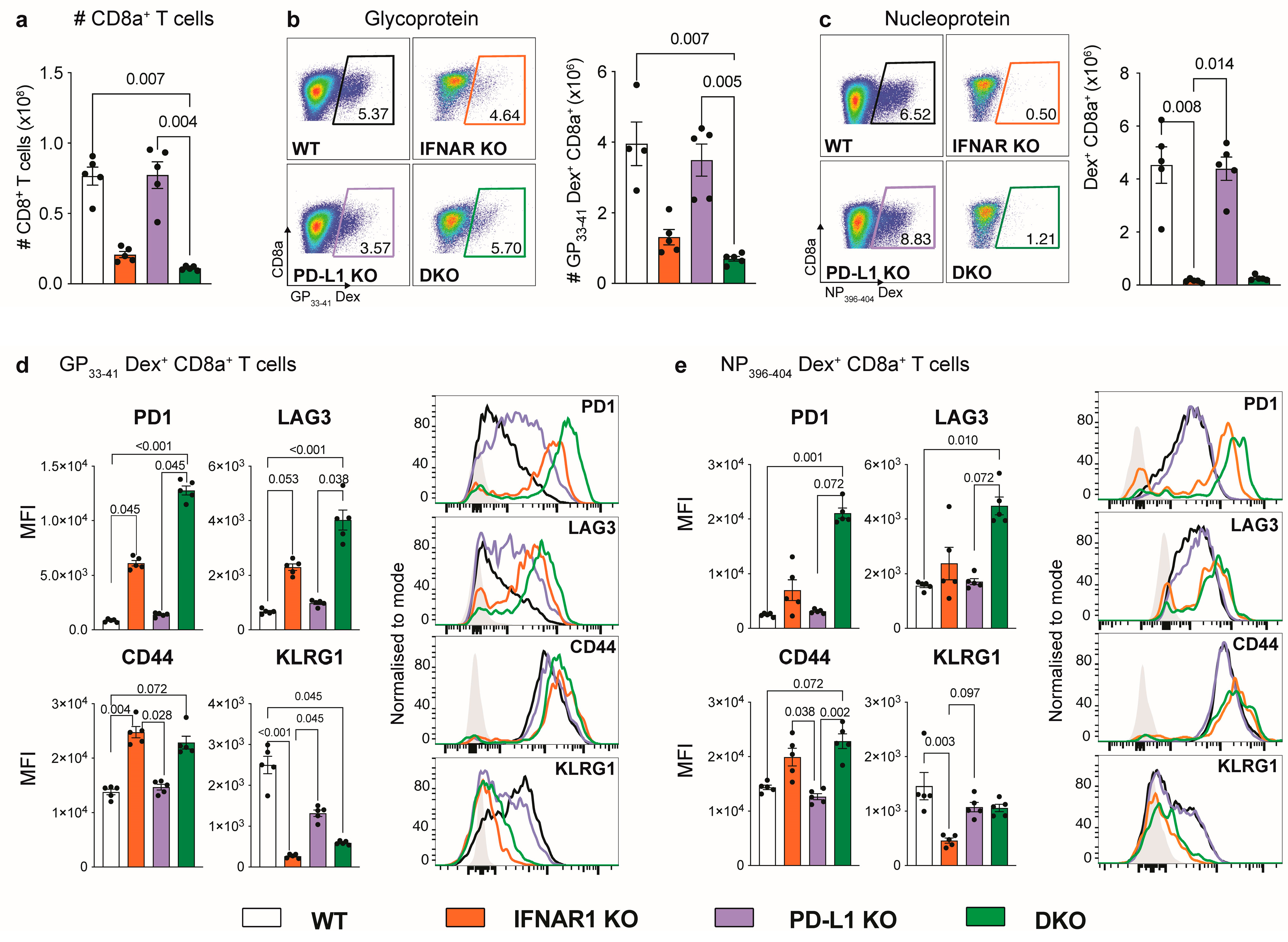
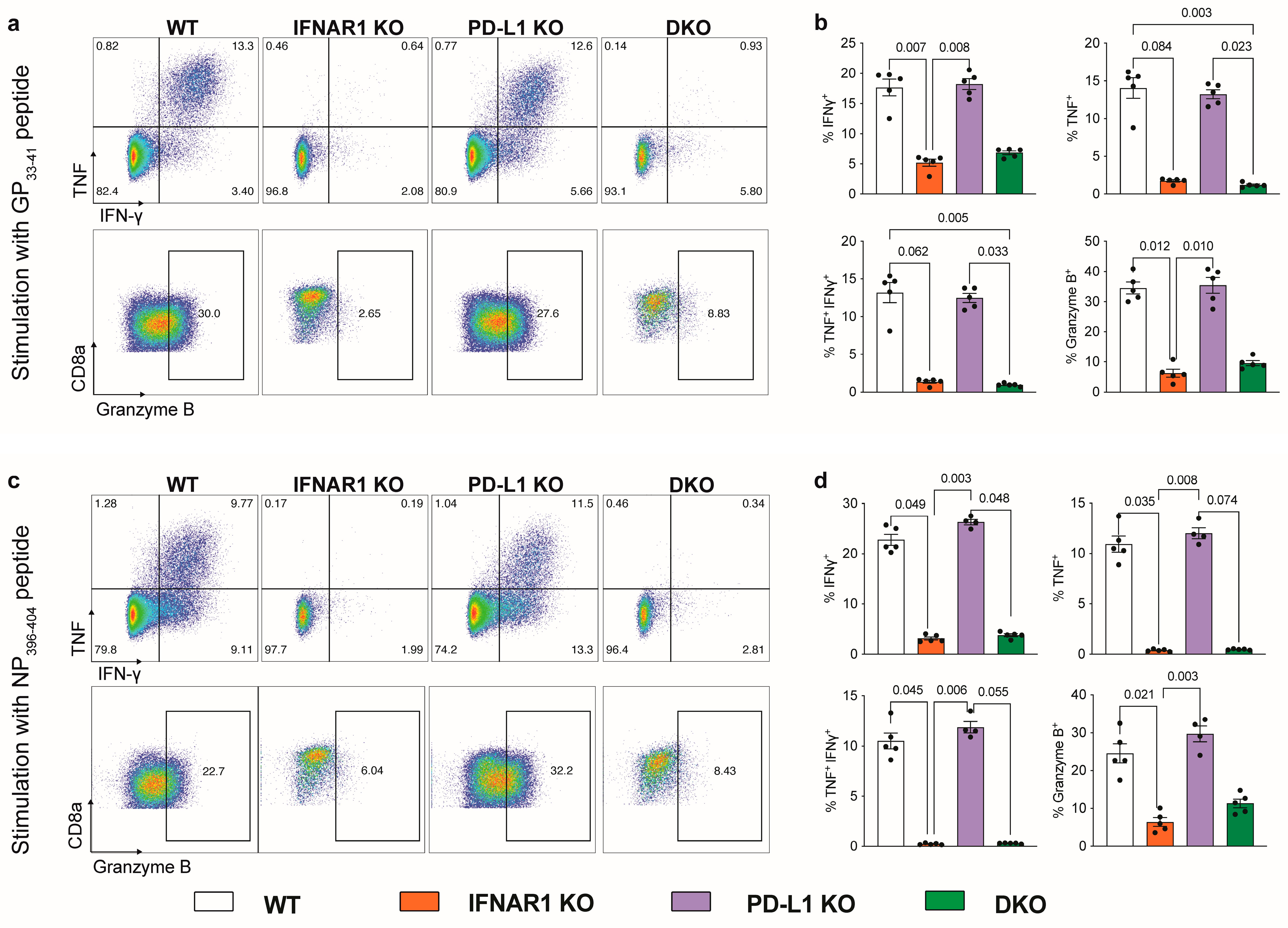
3.2. IFNAR1-Deficient Mice Fail to Accumulate LCMV-Specific CD8+ T Cells Independently of PD-L1
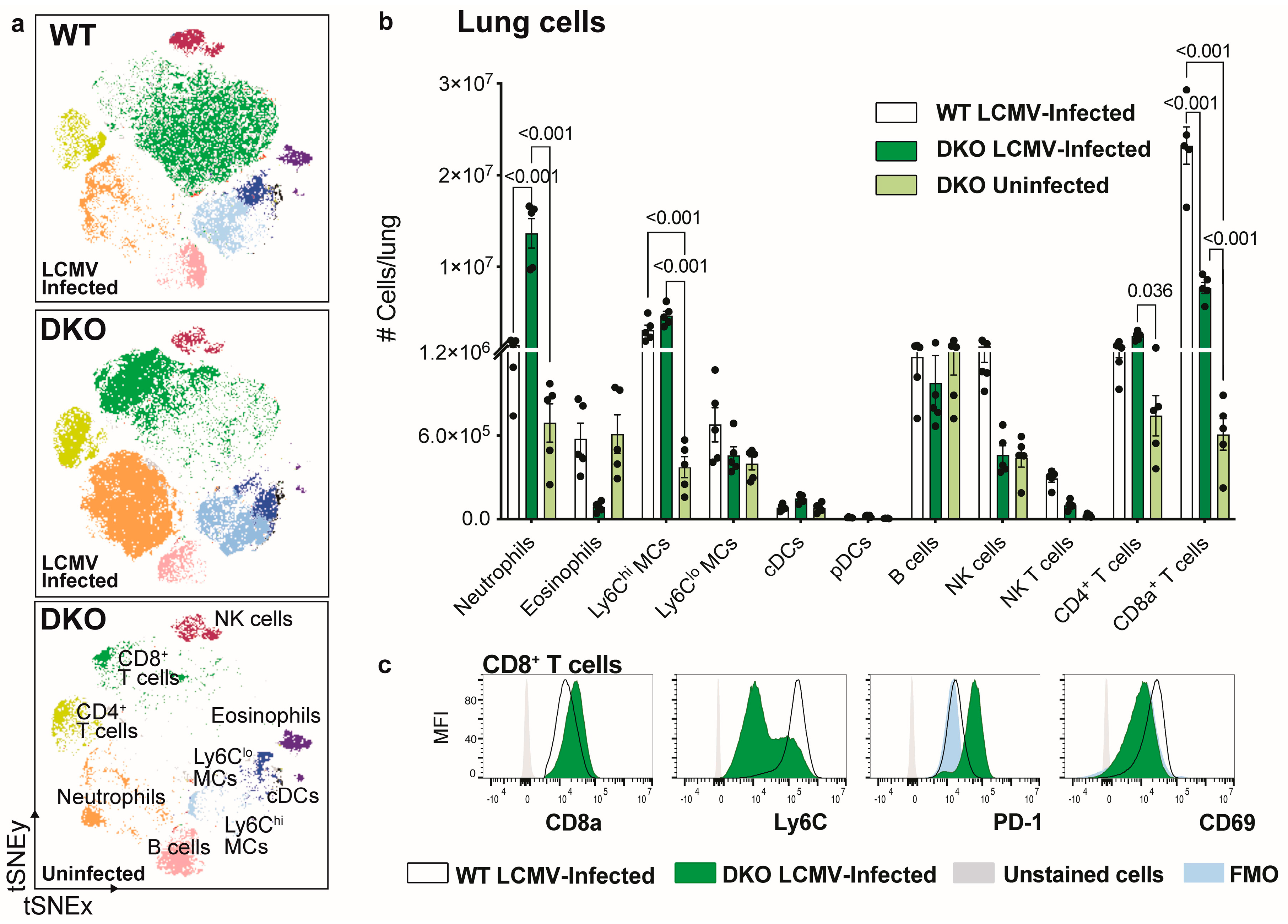
3.3. Ablation of IFNAR1 and PD-L1 Results in Enhanced Lung Pathology Following LCMV Infection
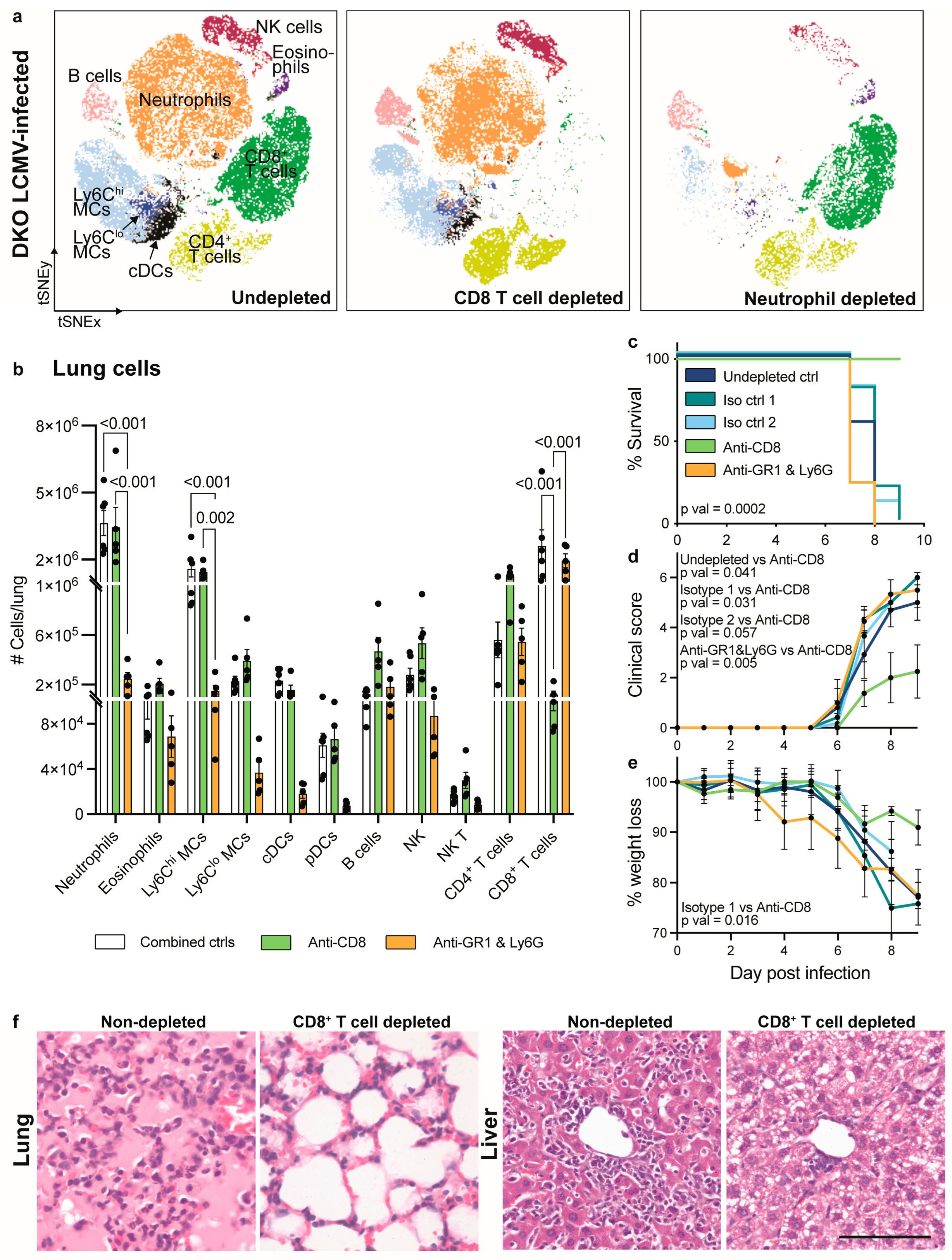
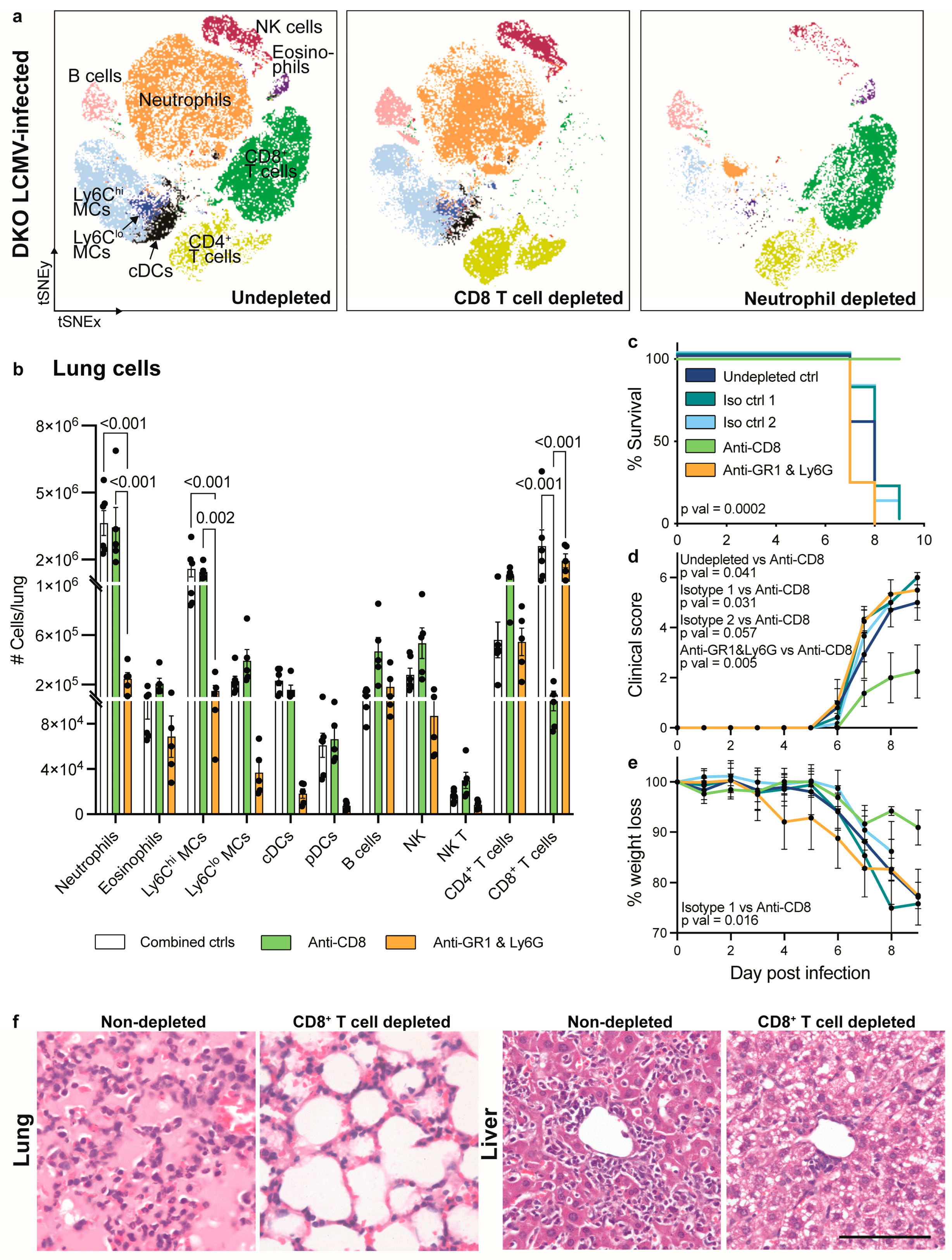
3.4. CD8+ T Cell Depletion but Not Neutrophil Depletion Rescues IFNAR1 x PD-L1 DKO Mice from Lethal LCMV Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suprunenko, T.; Hofer, M.J. Complexities of Type I Interferon Biology: Lessons from LCMV. Viruses 2019, 11, 172. [Google Scholar] [CrossRef]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Ou, R.; Zhou, S.; Huang, L.; Moskophidis, D. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J. Virol. 2001, 75, 8407–8423. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754. [Google Scholar] [CrossRef]
- Huber, M.; Suprunenko, T.; Ashhurst, T.; Marbach, F.; Raifer, H.; Wolff, S.; Strecker, T.; Viengkhou, B.; Jung, S.R.; Obermann, H.L.; et al. IRF9 Prevents CD8+ T Cell Exhaustion in an Extrinsic Manner during Acute Lymphocytic Choriomeningitis Virus Infection. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef]
- Keppler, S.J.; Rosenits, K.; Koegl, T.; Vucikuja, S.; Aichele, P. Signal 3 cytokines as modulators of primary immune responses during infections: The interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS ONE 2012, 7, e40865. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Van Pesch, V.; Lanaya, H.; Renauld, J.C.; Michiels, T. Characterization of the murine alpha interferon gene family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ahn, E.; Kissick, H.T.; Ahmed, R. Reinvigorating Exhausted T Cells by Blockade of the PD-1 Pathway. For. Immunopathol. Dis. Therap. 2015, 6, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Kornepati, A.V.R.; Vadlamudi, R.K.; Curiel, T.J. Programmed death ligand 1 signals in cancer cells. Nat. Rev. Cancer 2022, 22, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Upadhaya, S.; Neftelinov, S.T.; Hodge, J.; Campbell, J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat. Rev. Drug Discov. 2022, 21, 482–483. [Google Scholar] [CrossRef]
- Dutko, F.J.; Oldstone, M.B. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 1983, 64, 1689–1698. [Google Scholar] [CrossRef]
- Hofer, M.J.; Li, W.; Manders, P.; Terry, R.; Lim, S.L.; King, N.J.; Campbell, I.L. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J. Virol. 2012, 86, 6932–6946. [Google Scholar] [CrossRef]
- Asensio, V.C.; Campbell, I.L. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 1997, 71, 7832–7840. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Ashhurst, T.M.; Marsh-Wakefield, F.; Putri, G.H.; Spiteri, A.G.; Shinko, D.; Read, M.N.; Smith, A.L.; King, N.J.C. Integration, exploration, and analysis of high-dimensional single-cell cytometry data using Spectre. Cytometry A 2021, 101, 237–253. [Google Scholar] [CrossRef]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984, 160, 521–540. [Google Scholar] [CrossRef]
- Ashley, C.L.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Interferon-Independent Innate Responses to Cytomegalovirus. Front. Immunol. 2019, 10, 2751. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.L.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression. Viruses 2019, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.D.; Shin, H.; Kim, S.; Kim, H.J. Insights on interferon-independent induction of interferon-stimulated genes shaping the lung’s response in early SARS-CoV-2 infection. Heliyon 2023, 9, e22997. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.; Verbaro, D.J.; Egawa, T. PD-1 Signaling Promotes Control of Chronic Viral Infection by Restricting Type-I-Interferon-Mediated Tissue Damage. Cell Rep. 2019, 29, 2556–2564.e2553. [Google Scholar] [CrossRef]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef]
- Jennings, R.N.; Grayson, J.M.; Barton, E.S. Type I interferon signaling enhances CD8+ T cell effector function and differentiation during murine gammaherpesvirus 68 infection. J. Virol. 2014, 88, 14040–14049. [Google Scholar] [CrossRef]
- Speiser, D.E.; Utzschneider, D.T.; Oberle, S.G.; Munz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef]
- Frebel, H.; Nindl, V.; Schuepbach, R.A.; Braunschweiler, T.; Richter, K.; Vogel, J.; Wagner, C.A.; Loffing-Cueni, D.; Kurrer, M.; Ludewig, B.; et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med. 2012, 209, 2485–2499. [Google Scholar] [CrossRef]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Reichel, C.A.; Rehberg, M.; Lerchenberger, M.; Berberich, N.; Bihari, P.; Khandoga, A.G.; Zahler, S.; Krombach, F. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Hwaiz, R.; Rahman, M.; Syk, I.; Zhang, E.; Thorlacius, H. Rac1-dependent secretion of platelet-derived CCL5 regulates neutrophil recruitment via activation of alveolar macrophages in septic lung injury. J. Leukoc. Biol. 2015, 97, 975–984. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiteri, A.G.; Suprunenko, T.; Cutts, E.; Suen, A.; Ashhurst, T.M.; Viengkhou, B.; King, N.J.C.; Hofer, M.J. CD8+ T Cells Mediate Lethal Lung Pathology in the Absence of PD-L1 and Type I Interferon Signalling following LCMV Infection. Viruses 2024, 16, 390. https://doi.org/10.3390/v16030390
Spiteri AG, Suprunenko T, Cutts E, Suen A, Ashhurst TM, Viengkhou B, King NJC, Hofer MJ. CD8+ T Cells Mediate Lethal Lung Pathology in the Absence of PD-L1 and Type I Interferon Signalling following LCMV Infection. Viruses. 2024; 16(3):390. https://doi.org/10.3390/v16030390
Chicago/Turabian StyleSpiteri, Alanna G., Tamara Suprunenko, Erin Cutts, Andrew Suen, Thomas M. Ashhurst, Barney Viengkhou, Nicholas J. C. King, and Markus J. Hofer. 2024. "CD8+ T Cells Mediate Lethal Lung Pathology in the Absence of PD-L1 and Type I Interferon Signalling following LCMV Infection" Viruses 16, no. 3: 390. https://doi.org/10.3390/v16030390
APA StyleSpiteri, A. G., Suprunenko, T., Cutts, E., Suen, A., Ashhurst, T. M., Viengkhou, B., King, N. J. C., & Hofer, M. J. (2024). CD8+ T Cells Mediate Lethal Lung Pathology in the Absence of PD-L1 and Type I Interferon Signalling following LCMV Infection. Viruses, 16(3), 390. https://doi.org/10.3390/v16030390