
Quantification of Hepatitis E Virus ORF2 Protein by a Novel Sandwich ELISA
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Plasma Specimens
2.2. Animal Studies
2.3. Preparation of Recombinant ORF2 Antigens
2.4. Preparation of Mouse Monoclonal Antibodies against the ORF2 Antigen
2.5. Sandwich ELISA Protocol Development
2.6. Screening of Matched Antibody Pairs
2.7. Sandwich ELISA Protocol Optimization
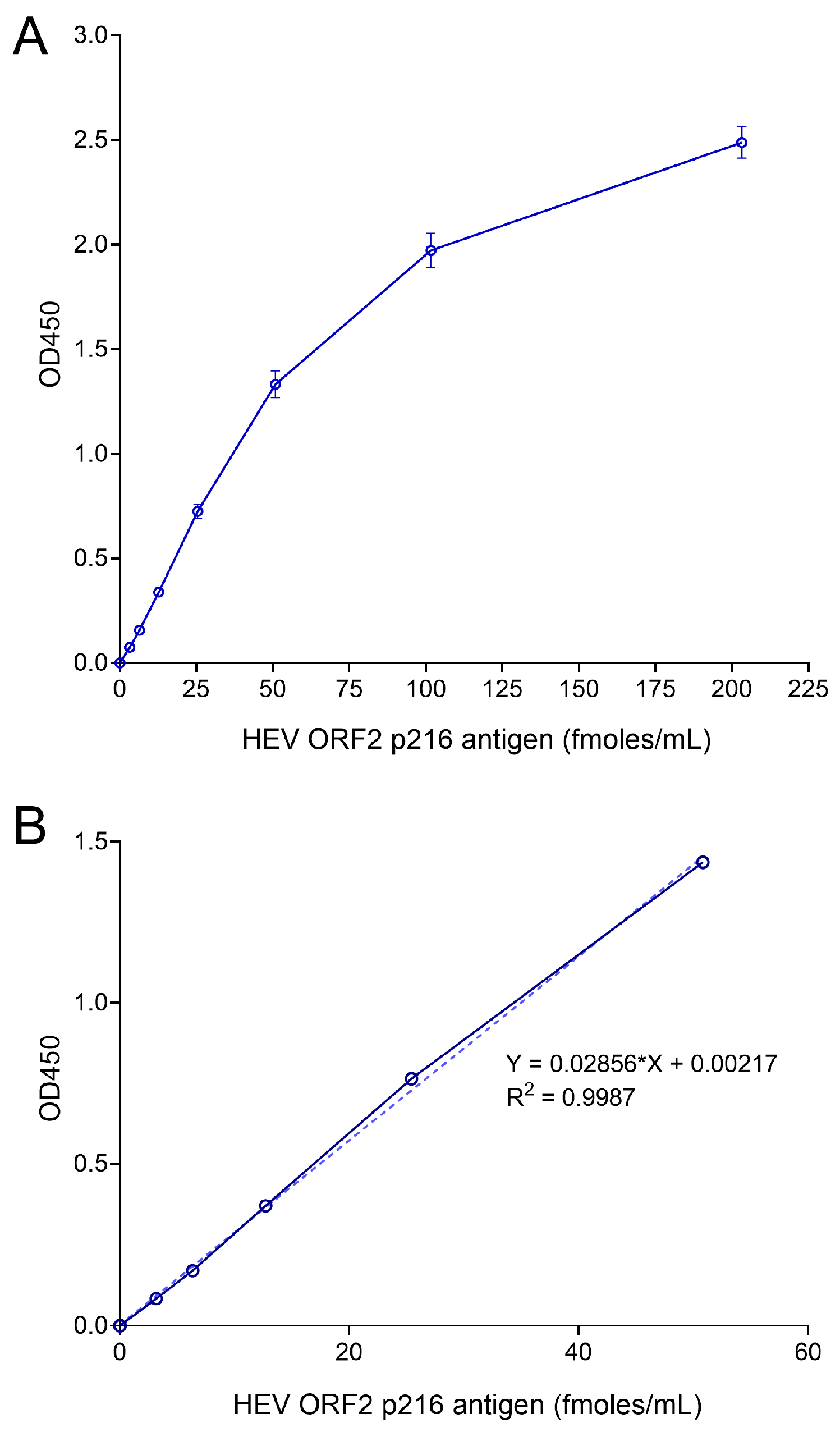
2.8. Generation of Standard Curves
2.9. Cross-Reactivity Analysis
2.10. HEV RNA Preparation and Transfection in Huh7 Cells
2.11. HEV Infection in Human Hepatocytes
2.12. HEV RT-qPCR
2.13. Quantification of ORF2 Antigen in Gerbil Serum Samples
2.14. Preparation of ORF2 Antigen-Spiked Human Plasma Samples
3. Results
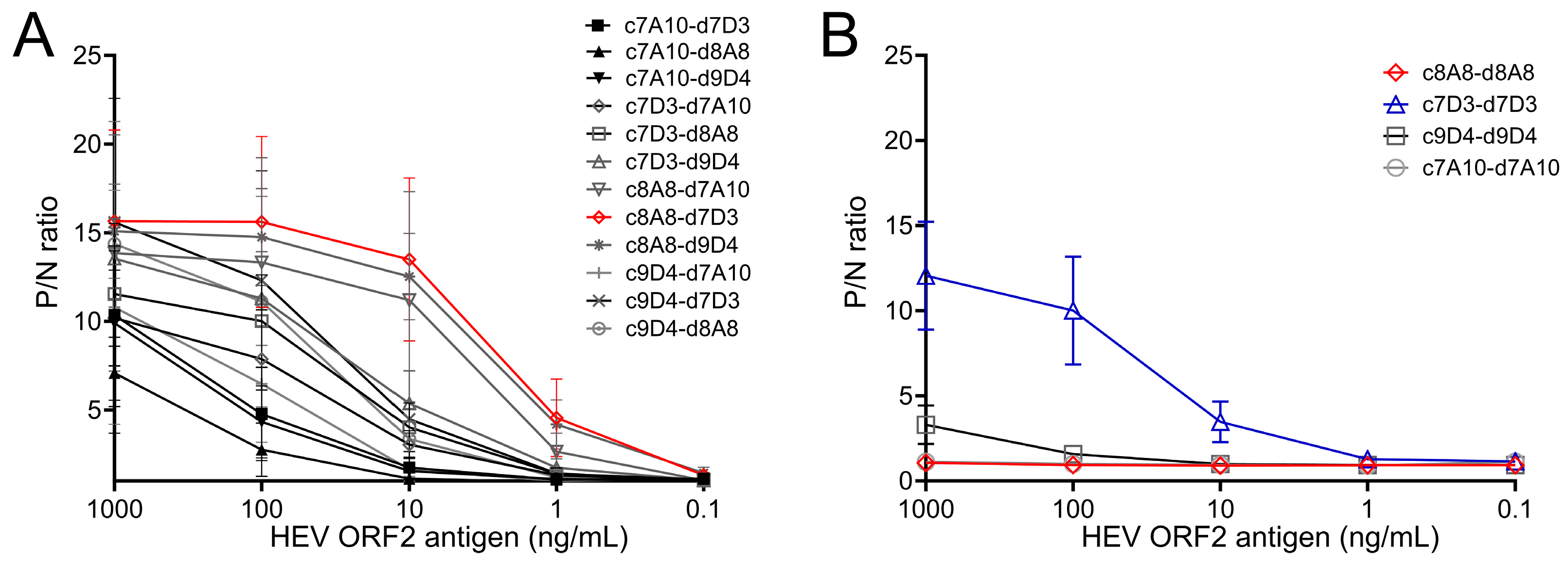
3.1. Selection of Monoclonal Antibodies for HEV ORF2 Antigen Sandwich ELISA
3.2. Optimization of Sandwich ELISA to Increase the Sensitivity of ORF2 Antigen Detection
3.3. Sensitivity and Specificity of the Sandwich ELISA for ORF2 Antigen of Different HEV Genotypes
3.4. Quantification of ORF2 Antigen in HEV-Infected Hepatocyte Culture Medium
3.5. Quantification of ORF2 Antigen in Sera of Gerbils Infected with HEV
3.6. Quantification of HEV ORF2 Antigen in Human Plasma Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Hepatitis Report 2017; License: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2017.
- Purdy, M.A.; Drexler, J.F.; Meng, X.J.; Norder, H.; Okamoto, H.; Van der Poel, W.H.M.; Reuter, G.; de Souza, W.M.; Ulrich, R.G.; Smith, D.B. ICTV Virus Taxonomy Profile: Hepeviridae 2022. J. Gen. Virol. 2022, 103, 001778. [Google Scholar] [CrossRef]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gracia, M.T.; Garcia, M.; Suay, B.; Mateos-Lindemann, M.L. Current Knowledge on Hepatitis E. J. Clin. Transl. Hepatol. 2015, 3, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Capai, L.; Charrel, R.; Falchi, A. Hepatitis E in High-Income Countries: What Do We Know? And What Are the Knowledge Gaps? Viruses 2018, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Legrand-Abravanel, F.; Kamar, N.; Izopet, J. Screening, diagnosis and risks associated with Hepatitis E virus infection. Expert Rev. Anti-Infect. Ther. 2019, 17, 403–418. [Google Scholar] [CrossRef]
- Norder, H.; Karlsson, M.; Mellgren, A.; Konar, J.; Sandberg, E.; Lasson, A.; Castedal, M.; Magnius, L.; Lagging, M. Diagnostic Performance of Five Assays for Anti-Hepatitis E Virus IgG and IgM in a Large Cohort Study. J. Clin. Microbiol. 2016, 54, 549–555. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis E virus infection. J. Hepatol. 2018, 68, 1256–1271. [Google Scholar] [CrossRef]
- Sakata, H.; Matsubayashi, K.; Iida, J.; Nakauchi, K.; Kishimoto, S.; Sato, S.; Ikuta, K.; Satake, M.; Kino, S. Trends in hepatitis E virus infection: Analyses of the long-term screening of blood donors in Hokkaido, Japan, 2005–2019. Transfusion 2021, 61, 3390–3401. [Google Scholar] [CrossRef]
- Boland, F.; Martinez, A.; Pomeroy, L.; O’Flaherty, N. Blood Donor Screening for Hepatitis E Virus in the European Union. Transfus. Med. Hemother 2019, 46, 95–103. [Google Scholar] [CrossRef]
- Talapko, J.; Mestrovic, T.; Pustijanac, E.; Skrlec, I. Towards the Improved Accuracy of Hepatitis E Diagnosis in Vulnerable and Target Groups: A Global Perspective on the Current State of Knowledge and the Implications for Practice. Healthcare 2021, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Lytton, S.D.; Bulbul, M.R.H.; Barua, K.; Begum, M.C.; Chowdhury, B.; Islam, Z.; Faiyaz, K.I.; Khan Chandan, M.S.; Shakeel, A.; Landt, O.; et al. Hepatitis E Virus Capsid Antigen (HEV-Ag)—A practical diagnostic biomarker in the HEV outbreak scenario. J. Clin. Virol. 2021, 134, 104692. [Google Scholar] [CrossRef]
- Wen, G.P.; Tang, Z.M.; Yang, F.; Zhang, K.; Ji, W.F.; Cai, W.; Huang, S.J.; Wu, T.; Zhang, J.; Zheng, Z.Z.; et al. A valuable antigen detection method for diagnosis of acute hepatitis E. J. Clin. Microbiol. 2015, 53, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Barciela, M.; Rando-Segura, A.; Barreira-Díaz, A.; Bes, M.; Ruzo, S.P.; Piron, M.; Quer, J.; Sauleda, S.; Rodríguez-Frías, F.; Esteban, R.; et al. Unexpected long-lasting anti-HEV IgM positivity: Is HEV antigen a better serological marker for hepatitis E infection diagnosis? J. Viral. Hepat. 2020, 27, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Tremeaux, P.; Lhomme, S.; Chapuy-Regaud, S.; Peron, J.M.; Alric, L.; Kamar, N.; Izopet, J.; Abravanel, F. Performance of an antigen assay for diagnosing acute hepatitis E virus genotype 3 infection. J. Clin. Virol. 2016, 79, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, P.; Bremer, B.; Todt, D.; Brown, R.J.; Heim, A.; Manns, M.P.; Steinmann, E.; Wedemeyer, H. Hepatitis E Virus (HEV) ORF2 Antigen Levels Differentiate between Acute and Chronic HEV Infection. J. Infect. Dis. 2016, 214, 361–368. [Google Scholar] [CrossRef]
- El-Mokhtar, M.A.; Karam-Allah Ramadan, H.; Abdel Hameed, M.R.; Kamel, A.M.; Mandour, S.A.; Ali, M.; Abdel-Malek, M.A.; Abd El-Kareem, D.M.; Adel, S.; Salama, E.H.; et al. Evaluation of hepatitis E antigen kinetics and its diagnostic utility for prediction of the outcomes of hepatitis E virus genotype 1 infection. Virulence 2021, 12, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Huang, Y.; Wang, P.; Li, Q.; Li, Z.; Jiang, J.; Guo, Q.; Gui, H.; Xie, Q. Dynamics of Hepatitis E Virus (HEV) Antibodies and Development of a Multifactorial Model To Improve the Diagnosis of HEV Infection in Resource-Limited Settings. J. Clin. Microbiol. 2021, 59, e02321-20. [Google Scholar] [CrossRef] [PubMed]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223.e218. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef]
- Ankavay, M.; Montpellier, C.; Sayed, I.M.; Saliou, J.M.; Wychowski, C.; Saas, L.; Duvet, S.; Aliouat-Denis, C.M.; Farhat, R.; de Masson d’Autume, V.; et al. New insights into the ORF2 capsid protein, a key player of the hepatitis E virus lifecycle. Sci. Rep. 2019, 9, 6243. [Google Scholar] [CrossRef]
- Fares-Gusmao, R.; Jiang, Z.; Subramaniam, S.; Visser, B.J.; Scott, A.; Ishida, Y.; Saito, T.; Baylis, S.A.; McGivern, D.R.; HEV Standard Calibration Study Group. Development and characterization of secondary standards for nucleic acid amplification technology (NAAT) assays for detection of hepatitis E virus. J. Clin. Virol. 2022, 157, 105325. [Google Scholar] [CrossRef] [PubMed]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Garson, J.A.; Ferns, R.B.; Grant, P.R.; Ijaz, S.; Nastouli, E.; Szypulska, R.; Tedder, R.S. Minor groove binder modification of widely used TaqMan probe for hepatitis E virus reduces risk of false negative real-time PCR results. J. Virol. Methods 2012, 186, 157–160. [Google Scholar] [CrossRef]
- Subramaniam, S.; Fares-Gusmao, R.; Sato, S.; Cullen, J.M.; Takeda, K.; Farci, P.; McGivern, D.R. Distinct disease features of acute and persistent genotype 3 hepatitis E virus infection in immunocompetent and immunosuppressed Mongolian gerbils. PLoS Pathog. 2023, 19, e1011664. [Google Scholar] [CrossRef]
- Vollmer, T.; Knabbe, C.; Dreier, J. Comparison of real-time PCR and antigen assays for detection of hepatitis E virus in blood donors. J. Clin. Microbiol. 2014, 52, 2150–2156. [Google Scholar] [CrossRef]
- Zhang, F.; Li, X.; Li, Z.; Harrison, T.J.; Chong, H.; Qiao, S.; Huang, W.; Zhang, H.; Zhuang, H.; Wang, Y. Detection of HEV antigen as a novel marker for the diagnosis of hepatitis E. J. Med. Virol. 2006, 78, 1441–1448. [Google Scholar] [CrossRef]
- Grandien, M. Viral diagnosis by antigen detection techniques. Clin. Diagn. Virol. 1996, 5, 81–90. [Google Scholar] [CrossRef]
- He, Q.; Zhang, F.; Shu, J.; Li, S.; Liang, Z.; Du, M.; Liu, X.; Liu, T.; Li, M.; Yin, X.; et al. Immunocompromised rabbit model of chronic HEV reveals liver fibrosis and distinct efficacy of different vaccination strategies. Hepatology 2022, 76, 788–802. [Google Scholar] [CrossRef]
- Li, S.; Tang, X.; Seetharaman, J.; Yang, C.; Gu, Y.; Zhang, J.; Du, H.; Shih, J.W.; Hew, C.L.; Sivaraman, J.; et al. Dimerization of hepatitis E virus capsid protein E2s domain is essential for virus-host interaction. PLoS Pathog. 2009, 5, e1000537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, X.J.; Tang, Z.M.; Yang, F.; Wang, S.L.; Cai, W.; Zhang, K.; Xia, N.S.; Zheng, Z.Z. A Comprehensive Study of Neutralizing Antigenic Sites on the Hepatitis E Virus (HEV) Capsid by Constructing, Clustering, and Characterizing a Tool Box. J. Biol. Chem. 2015, 290, 19910–19922. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, M.; Lin, Z.; Pan, H.; Tang, Z.; Zheng, Z.; Li, S.; Zhang, J.; Xia, N.; Zhao, Q. Multifaceted characterization of recombinant protein-based vaccines: An immunochemical toolbox for epitope-specific analyses of the hepatitis E vaccine. Vaccine 2018, 36, 7650–7658. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Zhang, Y.J. Molecular Biology and Infection of Hepatitis E Virus. Front. Microbiol. 2016, 7, 1419. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Behloul, N.; Wen, J.; Zhang, J.; Meng, J. Role of asparagine at position 562 in dimerization and immunogenicity of the hepatitis E virus capsid protein. Infect. Genet. Evol. 2016, 37, 99–107. [Google Scholar] [CrossRef]
- Ying, D.; He, Q.; Tian, W.; Chen, Y.; Zhang, X.; Wang, S.; Liu, C.; Chen, Z.; Liu, Y.; Fu, L.; et al. Urine is a viral antigen reservoir in hepatitis E virus infection. Hepatology 2023, 77, 1722–1734. [Google Scholar] [CrossRef]
- Li, T.C.; Bai, H.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Doan, Y.H.; Takahashi, K.; Mishiro, S.; Takeda, N.; Wakita, T. Genotype 5 Hepatitis E Virus Produced by a Reverse Genetics System Has the Potential for Zoonotic Infection. Hepatol. Commun. 2019, 3, 160–172. [Google Scholar] [CrossRef]
- Meister, T.L.; Bruggemann, Y.; Nocke, M.K.; Ulrich, R.G.; Schuhenn, J.; Sutter, K.; Gomer, A.; Bader, V.; Winklhofer, K.F.; Broering, R.; et al. A ribavirin-induced ORF2 single-nucleotide variant produces defective hepatitis E virus particles with immune decoy function. Proc. Natl. Acad. Sci. USA 2022, 119, e2202653119. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Sun, L.; Wang, Y.; Gao, S.; Yang, W.; Li, M. Different mutations at position 562 of the hepatitis E virus capsid proteins exhibit differential effects on viral neutralizing activity. Exp. Ther. Med. 2021, 21, 110. [Google Scholar] [CrossRef]
- Marion, O.; Capelli, N.; Lhomme, S.; Dubois, M.; Pucelle, M.; Abravanel, F.; Kamar, N.; Izopet, J. Hepatitis E virus genotype 3 and capsid protein in the blood and urine of immunocompromised patients. J. Infect. 2019, 78, 232–240. [Google Scholar] [CrossRef]
- Zhao, C.; Geng, Y.; Harrison, T.J.; Huang, W.; Song, A.; Wang, Y. Evaluation of an antigen-capture EIA for the diagnosis of hepatitis E virus infection. J. Viral Hepat. 2015, 22, 957–963. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, S.; Fares-Gusmao, R.; McGivern, D.R. Quantification of Hepatitis E Virus ORF2 Protein by a Novel Sandwich ELISA. Viruses 2024, 16, 393. https://doi.org/10.3390/v16030393
Subramaniam S, Fares-Gusmao R, McGivern DR. Quantification of Hepatitis E Virus ORF2 Protein by a Novel Sandwich ELISA. Viruses. 2024; 16(3):393. https://doi.org/10.3390/v16030393
Chicago/Turabian StyleSubramaniam, Sakthivel, Rafaelle Fares-Gusmao, and David R. McGivern. 2024. "Quantification of Hepatitis E Virus ORF2 Protein by a Novel Sandwich ELISA" Viruses 16, no. 3: 393. https://doi.org/10.3390/v16030393
APA StyleSubramaniam, S., Fares-Gusmao, R., & McGivern, D. R. (2024). Quantification of Hepatitis E Virus ORF2 Protein by a Novel Sandwich ELISA. Viruses, 16(3), 393. https://doi.org/10.3390/v16030393