
CRISPR RNA-Guided Transposases Facilitate Dispensable Gene Study in Phage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage, Strains, and Media
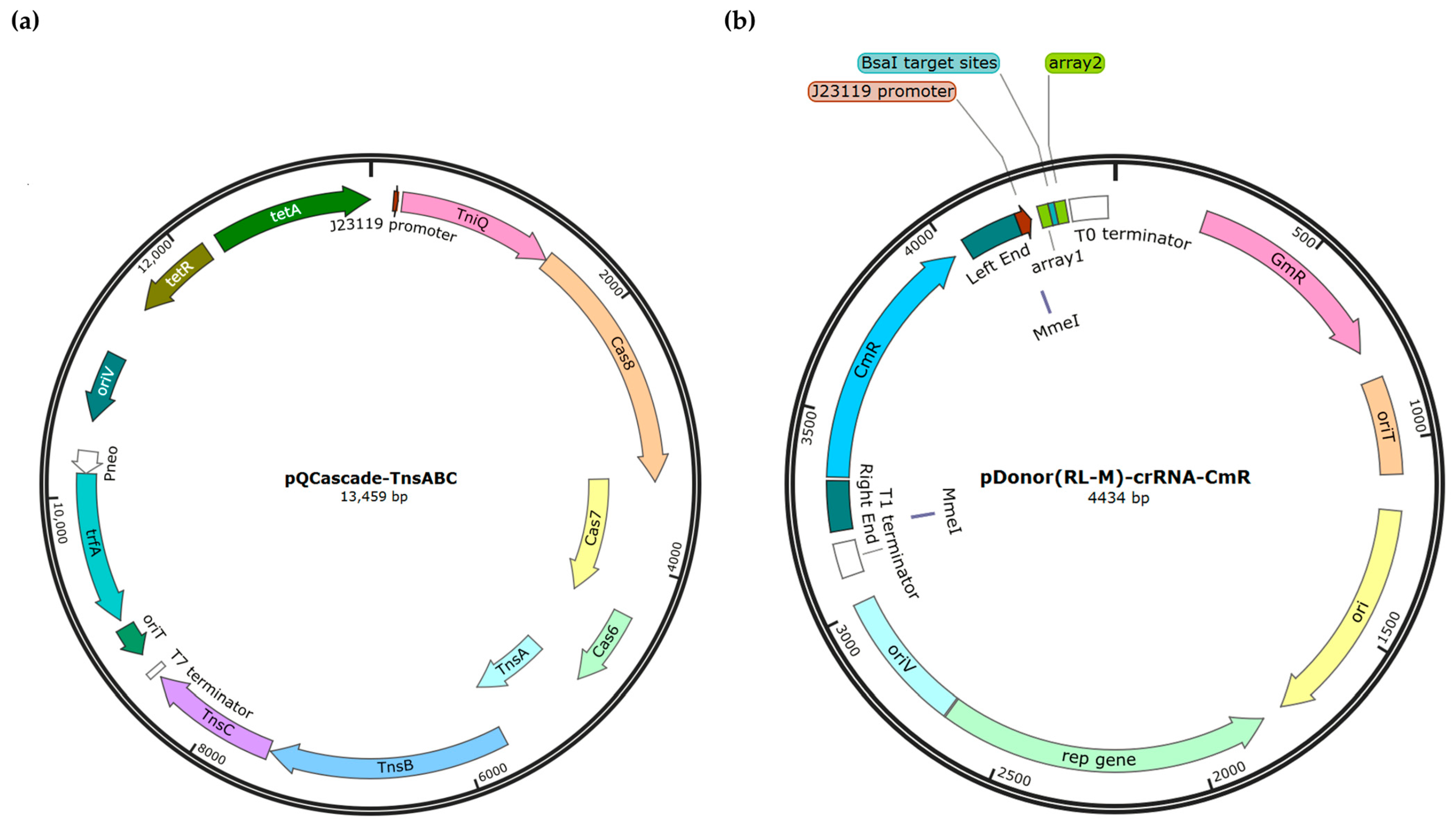
2.2. Plasmid Construction
2.3. Design of crRNA Spacers
2.4. Preparation P. aeruginosa PAO1 Cells for Transposon Insertion
2.5. Transposon Insertion Experiments
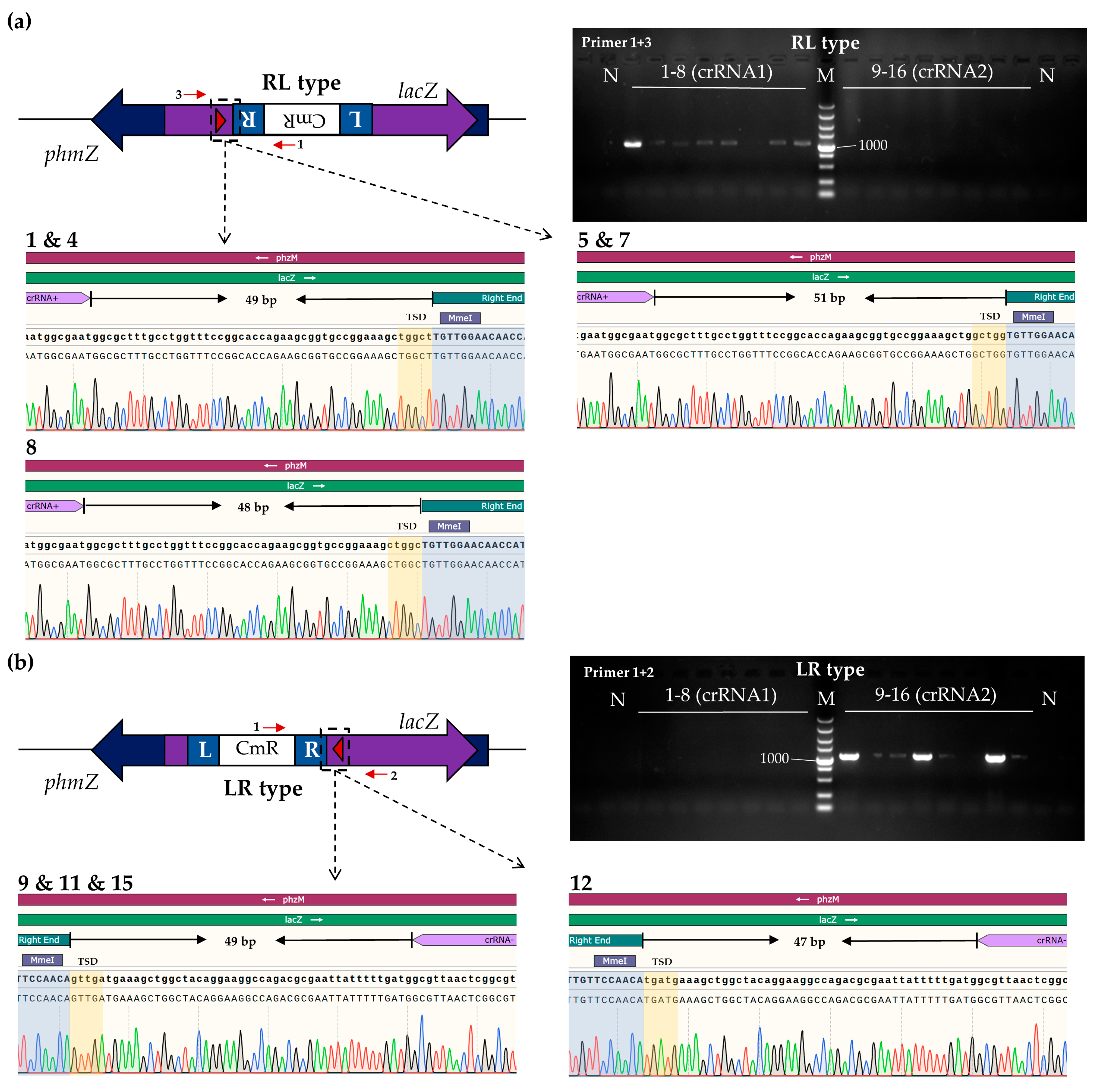
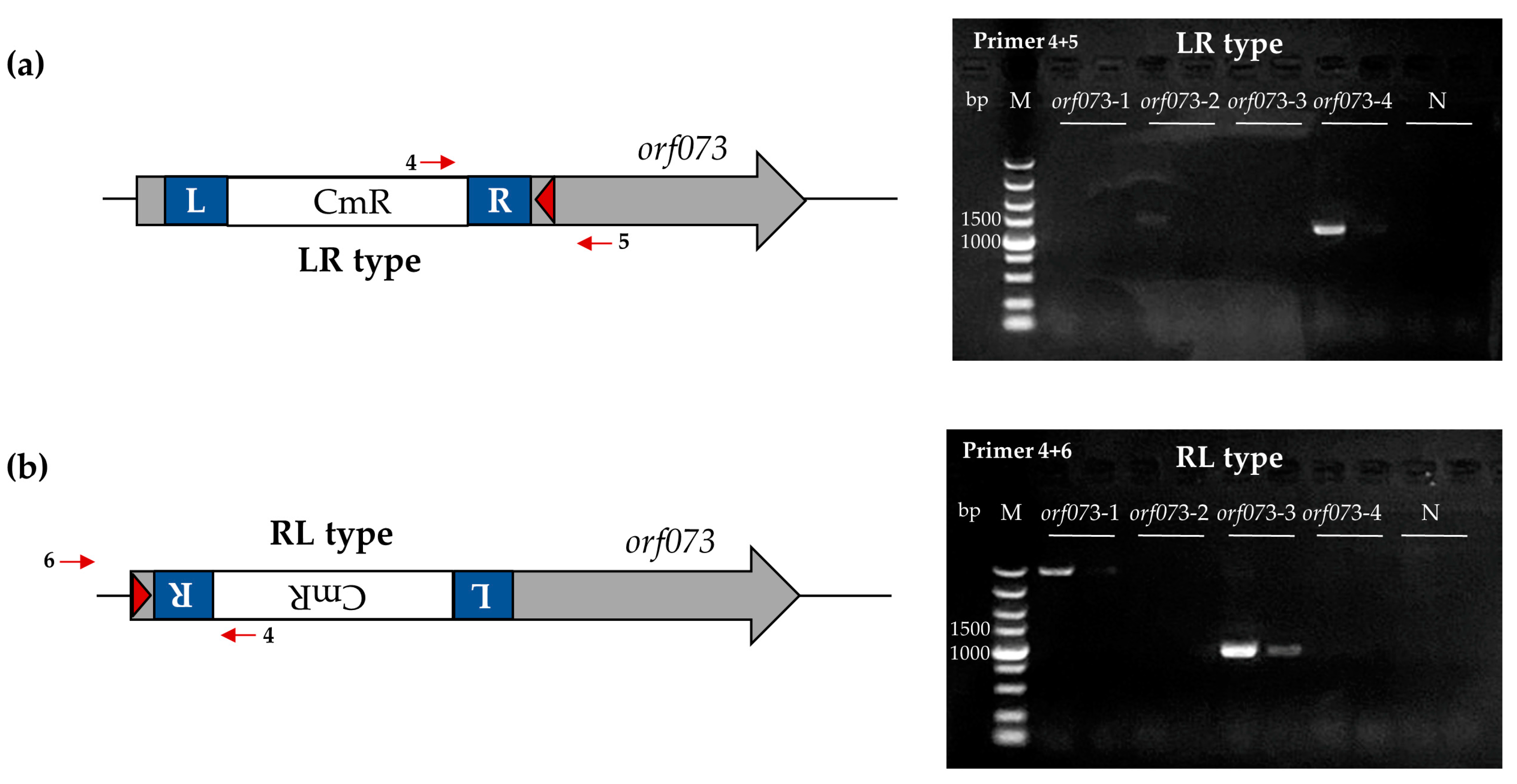
2.6. PCR and Sequencing Analysis of Transposon Insertion Products
2.7. Extraction of Phage Genomes
3. Results
3.1. Establishment of a Two-Plasmid Transposon Insertion System for P. aeruginosa Phage S1
3.2. Enrichment of Inserted Phages by AcrVA1
3.3. Transposon Insertions of Phage S1
3.4. Discovering Site-Specific Integration by Tn-Seq
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chevallereau, A.; Pons, B.J.; van Houte, S.; Westra, E.R. Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef]
- Pirnay, J.P.; Blasdel, B.G.; Bretaudeau, L.; Buckling, A.; Chanishvili, N.; Clark, J.R.; Corte-Real, S.; Debarbieux, L.; Dublanchet, A.; De Vos, D.; et al. Quality and safety requirements for sustainable phage therapy products. Pharm. Res. 2015, 32, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J. Designing P. aeruginosa synthetic phages with reduced genomes. Sci. Rep. 2021, 11, 2164. [Google Scholar] [CrossRef] [PubMed]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. MMBR 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Hatoum-Aslan, A. Phage Genetic Engineering Using CRISPR-Cas Systems. Viruses 2018, 10, 335. [Google Scholar] [CrossRef]
- Lemire, S.; Yehl, K.M.; Lu, T.K. Phage-Based Applications in Synthetic Biology. Annu. Rev. Virol. 2018, 5, 453–476. [Google Scholar] [CrossRef]
- Vo, P.L.H.; Ronda, C.; Klompe, S.E.; Chen, E.E.; Acree, C.; Wang, H.H.; Sternberg, S.H. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol. 2021, 39, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Qian, F.; Yang, J.; Liu, Y.; Dong, F.; Xu, C.; Sun, B.; Chen, B.; Xu, X.; Li, Y.; et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 2017, 8, 15179. [Google Scholar] [CrossRef] [PubMed]
- Vento, J.M.; Crook, N.; Beisel, C.L. Barriers to genome editing with CRISPR in bacteria. J. Ind. Microbiol. Biotechnol. 2019, 46, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Costa, A.R.; van Beljouw, S.P.B.; Fineran, P.C.; Brouns, S.J.J. Approaches for bacteriophage genome engineering. Trends Biotechnol. 2023, 41, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Kilcher, S.; Loessner, M.J. Engineering Bacteriophages as Versatile Biologics. Trends Microbiol. 2019, 27, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Kilcher, S.; Studer, P.; Muessner, C.; Klumpp, J.; Loessner, M.J. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.J.; Jia, P.P.; Yin, S.; Bu, L.K.; Yang, G.; Pei, D.S. Engineering bacteriophages for enhanced host range and efficacy: Insights from bacteriophage-bacteria interactions. Front. Microbiol. 2023, 14, 1172635. [Google Scholar] [CrossRef] [PubMed]
- Vilen, H.; Aalto, J.M.; Kassinen, A.; Paulin, L.; Savilahti, H. A direct transposon insertion tool for modification and functional analysis of viral genomes. J. Virol. 2003, 77, 123–134. [Google Scholar] [CrossRef]
- Kiljunen, S.; Vilen, H.; Pajunen, M.; Savilahti, H.; Skurnik, M. Nonessential genes of phage phiYeO3-12 include genes involved in adaptation to growth on Yersinia enterocolitica serotype O:3. J. Bacteriol. 2005, 187, 1405–1414. [Google Scholar] [CrossRef]
- Krupovic, M.; Vilen, H.; Bamford, J.K.; Kivelä, H.M.; Aalto, J.M.; Savilahti, H.; Bamford, D.H. Genome characterization of lipid-containing marine bacteriophage PM2 by transposon insertion mutagenesis. J. Virol. 2006, 80, 9270–9278. [Google Scholar] [CrossRef]
- Klompe, S.E.; Vo, P.L.H.; Halpin-Healy, T.S.; Sternberg, S.H. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 2019, 571, 219–225. [Google Scholar] [CrossRef]
- McDonald, N.D.; Regmi, A.; Morreale, D.P.; Borowski, J.D.; Boyd, E.F. CRISPR-Cas systems are present predominantly on mobile genetic elements in Vibrio species. BMC Genom. 2019, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Classification and Nomenclature of CRISPR-Cas Systems: Where from Here? CRISPR J. 2018, 1, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.E. Tn7. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, P.; Lin, Z.; Wang, T. Characterization of Two Pseudomonas aeruginosa Viruses vB_PaeM_SCUT-S1 and vB_PaeM_SCUT-S2. Viruses 2019, 11, 318. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef]
- Lin, Z.; Li, H.; He, L.; Jing, Y.; Pistolozzi, M.; Wang, T.; Ye, Y. Efficient genome editing for Pseudomonas aeruginosa using CRISPR-Cas12a. Gene 2021, 790, 145693. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Marino, N.D.; Zhang, J.Y.; Borges, A.L.; Sousa, A.A.; Leon, L.M.; Rauch, B.J.; Walton, R.T.; Berry, J.D.; Joung, J.K.; Kleinstiver, B.P.; et al. Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 2018, 362, 240–242. [Google Scholar] [CrossRef]
- Tao, P.; Wu, X.; Tang, W.C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952–1961. [Google Scholar] [CrossRef]
- David, W.; Russell, J.S. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Grigonyte, A.M.; Harrison, C.; MacDonald, P.R.; Montero-Blay, A.; Tridgett, M.; Duncan, J.; Sagona, A.P.; Constantinidou, C.; Jaramillo, A.; Millard, A. Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7. Viruses 2020, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Chamorro, L.; Boulanger, P.; Rossier, O. Strategies for Bacteriophage T5 Mutagenesis: Expanding the Toolbox for Phage Genome Engineering. Front. Microbiol. 2021, 12, 667332. [Google Scholar] [CrossRef] [PubMed]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Mayo-Muñoz, D.; He, F.; Jørgensen, J.B.; Madsen, P.K.; Bhoobalan-Chitty, Y.; Peng, X. Anti-CRISPR-Based and CRISPR-Based Genome Editing of Sulfolobus islandicus Rod-Shaped Virus 2. Viruses 2018, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- van Opijnen, T.; Bodi, K.L.; Camilli, A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 2009, 6, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Jariah, R.O.A.; Hakim, M.S. Interaction of phages, bacteria, and the human immune system: Evolutionary changes in phage therapy. Rev. Med. Virol. 2019, 29, e2055. [Google Scholar] [CrossRef]
- Cordes, C.; Meima, R.; Twiest, B.; Kazemier, B.; Venema, G.; van Dijl, J.M.; Bron, S. The expression of a plasmid-specified exported protein causes structural plasmid instability in Bacillus subtilis. J. Bacteriol. 1996, 178, 5235–5242. [Google Scholar] [CrossRef]
- Klompe, S.E.; Jaber, N.; Beh, L.Y.; Mohabir, J.T.; Bernheim, A.; Sternberg, S.H. Evolutionary and mechanistic diversity of Type I-F CRISPR-associated transposons. Mol. Cell 2022, 82, 616–628.e5. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liang, Z.; Yu, S.; Ye, Y.; Lin, Z. CRISPR RNA-Guided Transposases Facilitate Dispensable Gene Study in Phage. Viruses 2024, 16, 422. https://doi.org/10.3390/v16030422
Liu Y, Liang Z, Yu S, Ye Y, Lin Z. CRISPR RNA-Guided Transposases Facilitate Dispensable Gene Study in Phage. Viruses. 2024; 16(3):422. https://doi.org/10.3390/v16030422
Chicago/Turabian StyleLiu, Yanmei, Zizhen Liang, Shuting Yu, Yanrui Ye, and Zhanglin Lin. 2024. "CRISPR RNA-Guided Transposases Facilitate Dispensable Gene Study in Phage" Viruses 16, no. 3: 422. https://doi.org/10.3390/v16030422