
Natural Adeno-Associated Virus Serotypes and Engineered Adeno-Associated Virus Capsid Variants: Tropism Differences and Mechanistic Insights

 ,
,
Abstract
:1. Introduction
2. Discovery of AAV and Their Evolution as Gene Therapy Tools
3. Transduction Mechanisms of Natural AAV Serotypes
3.1. AAV Cell Surface Receptors
3.2. AAV Endocytosis
3.3. Intracellular Retrograde Trafficking
3.4. Endosomal Processing and Escape into the Cytosol
3.5. Nuclear Entry, Uncoating, and Second-Strand Synthesis
4. Novel AAV Variants for Targeted Tropism
4.1. Directed Evolution
4.1.1. Peptide Insertion for CNS Targeting
4.1.2. Peptide Insertion for PNS Targeting
4.1.3. Peptide Insertion for Eye or Ear Targeting
4.1.4. Peptide Insertion for Heart and Muscle Targeting
4.1.5. Peptide Insertion for Lung or Liver Targeting
4.1.6. Gene Shuffling
4.1.7. Error-Prone PCR
4.2. Rational Design
4.3. Ancestral Capsid Reconstruction
5. Mechanistic Insight for the Transduction of Novel AAV Capsid Variants
5.1. CNS Variants
5.2. Skeletal Muscle and Heart Variants
5.3. Lung and Liver Variants
6. Conclusions, Perspectives, and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bedbrook, C.N.; Deverman, B.E.; Gradinaru, V. Viral Strategies for Targeting the Central and Peripheral Nervous Systems. Annu. Rev. Neurosci. 2018, 41, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Buning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Campos, L.J.; Arokiaraj, C.M.; Chuapoco, M.R.; Chen, X.; Goeden, N.; Gradinaru, V.; Fox, A.S. Advances in AAV technology for delivering genetically encoded cargo to the nonhuman primate nervous system. Curr. Res. Neurobiol. 2023, 4, 100086. [Google Scholar] [CrossRef]
- Challis, R.C.; Ravindra Kumar, S.; Chen, X.; Goertsen, D.; Coughlin, G.M.; Hori, A.M.; Chuapoco, M.R.; Otis, T.S.; Miles, T.F.; Gradinaru, V. Adeno-Associated Virus Toolkit to Target Diverse Brain Cells. Annu. Rev. Neurosci. 2022, 45, 447–469. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Lisowski, L.; Tay, S.S.; Alexander, I.E. Adeno-associated virus serotypes for gene therapeutics. Curr. Opin. Pharmacol. 2015, 24, 59–67. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Mayor, H.D.; Jamison, R.M.; Jordan, L.E.; Melnick, J.L. Structure and composition of a small particle prepared from a simian adenovirus. J. Bacteriol. 1965, 90, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.L.; Mayor, H.D.; Smith, K.O.; Rapp, F. Association of 20-Millimicron Particles with Adenoviruses. J. Bacteriol. 1965, 90, 271–274. [Google Scholar] [CrossRef]
- Buller, R.M.; Janik, J.E.; Sebring, E.D.; Rose, J.A. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol. 1981, 40, 241–247. [Google Scholar] [CrossRef]
- Thomson, B.J.; Weindler, F.W.; Gray, D.; Schwaab, V.; Heilbronn, R. Human herpesvirus 6 (HHV-6) is a helper virus for adeno-associated virus type 2 (AAV-2) and the AAV-2 rep gene homologue in HHV-6 can mediate AAV-2 DNA replication and regulate gene expression. Virology 1994, 204, 304–311. [Google Scholar] [CrossRef]
- Schlehofer, J.R.; Ehrbar, M.; zur Hausen, H. Vaccinia virus, herpes simplex virus, and carcinogens induce DNA amplification in a human cell line and support replication of a helpervirus dependent parvovirus. Virology 1986, 152, 110–117. [Google Scholar] [CrossRef]
- Walz, C.; Deprez, A.; Dupressoir, T.; Dürst, M.; Rabreau, M.; Schlehofer, J.R. Interaction of human papillomavirus type 16 and adeno-associated virus type 2 co-infecting human cervical epithelium. J. Gen. Virol. 1997, 78 Pt 6, 1441–1452. [Google Scholar] [CrossRef]
- Srivastava, A.; Lusby, E.W.; Berns, K.I. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 1983, 45, 555–564. [Google Scholar] [CrossRef]
- Lusby, E.; Fife, K.H.; Berns, K.I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. J. Virol. 1980, 34, 402–409. [Google Scholar] [CrossRef]
- Carter, B.J.; Khoury, G.; Denhardt, D.T. Physical map and strand polarity of specific fragments of adenovirus-associated virus DNA produced by endonuclease R-EcoRI. J. Virol. 1975, 16, 559–568. [Google Scholar] [CrossRef]
- Carter, B.J.; Khoury, G.; Rose, J.A. Adenovirus-associated virus multiplication. IX. Extent of transcription of the viral genome in vivo. J. Virol. 1972, 10, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Koczot, F.J.; Carter, B.J.; Garon, C.F.; Rose, J.A. Self-complementarity of terminal sequences within plus or minus strands of adenovirus-associated virus DNA. Proc. Natl. Acad. Sci. USA 1973, 70, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.E.; Sebring, E.D.; Rose, J.A. Concatemers of alternating plus and minus strands are intermediates in adenovirus-associated virus DNA synthesis. Proc. Natl. Acad. Sci. USA 1976, 73, 742–746. [Google Scholar] [CrossRef]
- Laughlin, C.A.; Westphal, H.; Carter, B.J. Spliced adenovirus-associated virus RNA. Proc. Natl. Acad. Sci. USA 1979, 76, 5567–5571. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.J.; Laughlin, C.A.; Carter, B.J. Adeno-associated virus RNA transcription in vivo. Eur. J. Biochem. 1981, 121, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Balagúe, C.; Kalla, M.; Zhang, W.W. Adeno-associated virus Rep78 protein and terminal repeats enhance integration of DNA sequences into the cellular genome. J. Virol. 1997, 71, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Surosky, R.T.; Urabe, M.; Godwin, S.G.; McQuiston, S.A.; Kurtzman, G.J.; Ozawa, K.; Natsoulis, G. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J. Virol. 1997, 71, 7951–7959. [Google Scholar] [CrossRef] [PubMed]
- King, J.A.; Dubielzig, R.; Grimm, D.; Kleinschmidt, J.A. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 2001, 20, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.A.; Maizel, J.V., Jr.; Inman, J.K.; Shatkin, A.J. Structural proteins of adenovirus-associated viruses. J. Virol. 1971, 8, 766–770. [Google Scholar] [CrossRef]
- Snijder, J.; van de Waterbeemd, M.; Damoc, E.; Denisov, E.; Grinfeld, D.; Bennett, A.; Agbandje-McKenna, M.; Makarov, A.; Heck, A.J. Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by Orbitrap mass spectrometry. J. Am. Chem. Soc. 2014, 136, 7295–7299. [Google Scholar] [CrossRef]
- Johnson, F.B.; Ozer, H.L.; Hoggan, M.D. Structural proteins of adenovirus-associated virus type 3. J. Virol. 1971, 8, 860–863. [Google Scholar] [CrossRef]
- Kohlbrenner, E.; Aslanidi, G.; Nash, K.; Shklyaev, S.; Campbell-Thompson, M.; Byrne, B.J.; Snyder, R.O.; Muzyczka, N.; Warrington, K.H., Jr.; Zolotukhin, S. Successful production of pseudotyped rAAV vectors using a modified baculovirus expression system. Mol. Ther. 2005, 12, 1217–1225. [Google Scholar] [CrossRef]
- Trempe, J.P.; Carter, B.J. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J. Virol. 1988, 62, 3356–3363. [Google Scholar] [CrossRef]
- Cassinotti, P.; Weitz, M.; Tratschin, J.D. Organization of the adeno-associated virus (AAV) capsid gene: Mapping of a minor spliced mRNA coding for virus capsid protein 1. Virology 1988, 167, 176–184. [Google Scholar] [CrossRef]
- Becerra, S.P.; Rose, J.A.; Hardy, M.; Baroudy, B.M.; Anderson, C.W. Direct mapping of adeno-associated virus capsid proteins B and C: A possible ACG initiation codon. Proc. Natl. Acad. Sci. USA 1985, 82, 7919–7923. [Google Scholar] [CrossRef] [PubMed]
- Becerra, S.P.; Koczot, F.; Fabisch, P.; Rose, J.A. Synthesis of adeno-associated virus structural proteins requires both alternative mRNA splicing and alternative initiations from a single transcript. J. Virol. 1988, 62, 2745–2754. [Google Scholar] [CrossRef] [PubMed]
- Grieger, J.C.; Snowdy, S.; Samulski, R.J. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J. Virol. 2006, 80, 5199–5210. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.; Ishizu, K.; Matsumoto, A.; Han, S.I.; Arisaka, F.; Takayama, M.; Suzuki, K.; Kato, K.; Kanda, T.; Watanabe, H.; et al. Nuclear transport of the major capsid protein is essential for adeno-associated virus capsid formation. J. Virol. 1999, 73, 7912–7915. [Google Scholar] [CrossRef]
- Girod, A.; Wobus, C.E.; Zadori, Z.; Ried, M.; Leike, K.; Tijssen, P.; Kleinschmidt, J.A.; Hallek, M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 2002, 83, 973–978. [Google Scholar] [CrossRef]
- Sonntag, F.; Schmidt, K.; Kleinschmidt, J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef]
- Earley, L.F.; Powers, J.M.; Adachi, K.; Baumgart, J.T.; Meyer, N.L.; Xie, Q.; Chapman, M.S.; Nakai, H. Adeno-associated Virus (AAV) Assembly-Activating Protein Is Not an Essential Requirement for Capsid Assembly of AAV Serotypes 4, 5, and 11. J. Virol. 2017, 91, e01980-16. [Google Scholar] [CrossRef]
- Sonntag, F.; Kother, K.; Schmidt, K.; Weghofer, M.; Raupp, C.; Nieto, K.; Kuck, A.; Gerlach, B.; Bottcher, B.; Muller, O.J.; et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011, 85, 12686–12697. [Google Scholar] [CrossRef] [PubMed]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef]
- Galibert, L.; Hyvönen, A.; Eriksson, R.A.E.; Mattola, S.; Aho, V.; Salminen, S.; Albers, J.D.; Peltola, S.K.; Weman, S.; Nieminen, T.; et al. Functional roles of the membrane-associated AAV protein MAAP. Sci. Rep. 2021, 11, 21698. [Google Scholar] [CrossRef] [PubMed]
- Elmore, Z.C.; Patrick Havlik, L.; Oh, D.K.; Anderson, L.; Daaboul, G.; Devlin, G.W.; Vincent, H.A.; Asokan, A. The membrane associated accessory protein is an adeno-associated viral egress factor. Nat. Commun. 2021, 12, 6239. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef]
- Drouin, L.M.; Agbandje-McKenna, M. Adeno-associated virus structural biology as a tool in vector development. Future Virol. 2013, 8, 1183–1199. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Weber, T. Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe 2011, 10, 563–576. [Google Scholar] [CrossRef]
- Gurda, B.L.; Raupp, C.; Popa-Wagner, R.; Naumer, M.; Olson, N.H.; Ng, R.; McKenna, R.; Baker, T.S.; Kleinschmidt, J.A.; Agbandje-McKenna, M. Mapping a neutralizing epitope onto the capsid of adeno-associated virus serotype 8. J. Virol. 2012, 86, 7739–7751. [Google Scholar] [CrossRef]
- DiMattia, M.A.; Nam, H.J.; Van Vliet, K.; Mitchell, M.; Bennett, A.; Gurda, B.L.; McKenna, R.; Olson, N.H.; Sinkovits, R.S.; Potter, M.; et al. Structural insight into the unique properties of adeno-associated virus serotype 9. J. Virol. 2012, 86, 6947–6958. [Google Scholar] [CrossRef]
- Gurda, B.L.; DiMattia, M.A.; Miller, E.B.; Bennett, A.; McKenna, R.; Weichert, W.S.; Nelson, C.D.; Chen, W.J.; Muzyczka, N.; Olson, N.H.; et al. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J. Virol. 2013, 87, 9111–9124. [Google Scholar] [CrossRef]
- Nam, H.J.; Gurda, B.L.; McKenna, R.; Potter, M.; Byrne, B.; Salganik, M.; Muzyczka, N.; Agbandje-McKenna, M. Structural studies of adeno-associated virus serotype 8 capsid transitions associated with endosomal trafficking. J. Virol. 2011, 85, 11791–11799. [Google Scholar] [CrossRef]
- Ng, R.; Govindasamy, L.; Gurda, B.L.; McKenna, R.; Kozyreva, O.G.; Samulski, R.J.; Parent, K.N.; Baker, T.S.; Agbandje-McKenna, M. Structural characterization of the dual glycan binding adeno-associated virus serotype 6. J. Virol. 2010, 84, 12945–12957. [Google Scholar] [CrossRef] [PubMed]
- Govindasamy, L.; Padron, E.; McKenna, R.; Muzyczka, N.; Kaludov, N.; Chiorini, J.A.; Agbandje-McKenna, M. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J. Virol. 2006, 80, 11556–11570. [Google Scholar] [CrossRef]
- Xie, Q.; Bu, W.; Bhatia, S.; Hare, J.; Somasundaram, T.; Azzi, A.; Chapman, M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 10405–10410. [Google Scholar] [CrossRef]
- Mietzsch, M.; Jose, A.; Chipman, P.; Bhattacharya, N.; Daneshparvar, N.; McKenna, R.; Agbandje-McKenna, M. Completion of the AAV Structural Atlas: Serotype Capsid Structures Reveals Clade-Specific Features. Viruses 2021, 13, 101. [Google Scholar] [CrossRef]
- Bleker, S.; Pawlita, M.; Kleinschmidt, J.A. Impact of capsid conformation and Rep-capsid interactions on adeno-associated virus type 2 genome packaging. J. Virol. 2006, 80, 810–820. [Google Scholar] [CrossRef]
- Tseng, Y.S.; Agbandje-McKenna, M. Mapping the AAV Capsid Host Antibody Response toward the Development of Second Generation Gene Delivery Vectors. Front. Immunol. 2014, 5, 9. [Google Scholar] [CrossRef]
- Opie, S.R.; Warrington, K.H., Jr.; Agbandje-McKenna, M.; Zolotukhin, S.; Muzyczka, N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003, 77, 6995–7006. [Google Scholar] [CrossRef] [PubMed]
- Zinn, E.; Vandenberghe, L.H. Adeno-associated virus: Fit to serve. Curr. Opin. Virol. 2014, 8, 90–97. [Google Scholar] [CrossRef]
- Samulski, R.J.; Chang, L.S.; Shenk, T. A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J. Virol. 1987, 61, 3096–3101. [Google Scholar] [CrossRef]
- Xiao, X.; Xiao, W.; Li, J.; Samulski, R.J. A novel 165-base-pair terminal repeat sequence is the sole cis requirement for the adeno-associated virus life cycle. J. Virol. 1997, 71, 941–948. [Google Scholar] [CrossRef]
- Flotte, T.R.; Afione, S.A.; Conrad, C.; McGrath, S.A.; Solow, R.; Oka, H.; Zeitlin, P.L.; Guggino, W.B.; Carter, B.J. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA 1993, 90, 10613–10617. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Leone, P.; Samulski, R.J.; Xiao, X.; Pfaff, D.W.; O’Malley, K.L.; During, M.J. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 1994, 8, 148–154. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Xiao, X.; Samulski, R.J.; Li, J.; Ojamaa, K.; Klein, I.L.; Makimura, H.; Kaplitt, M.J.; Strumpf, R.K.; Diethrich, E.B. Long-term gene transfer in porcine myocardium after coronary infusion of an adeno-associated virus vector. Ann. Thorac. Surg. 1996, 62, 1669–1676. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Liu, Q.; Bowen, G.P.; Wong, N.C.; Bartlett, J.S.; Muruve, D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002, 76, 4580–4590. [Google Scholar] [CrossRef]
- Somanathan, S.; Breous, E.; Bell, P.; Wilson, J.M. AAV vectors avoid inflammatory signals necessary to render transduced hepatocyte targets for destructive T cells. Mol. Ther. 2010, 18, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.; Carter, B.; Conrad, C.; Guggino, W.; Reynolds, T.; Rosenstein, B.; Taylor, G.; Walden, S.; Wetzel, R. A phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung disease. Hum. Gene Ther. 1996, 7, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Gene Therapy Clinical Trials Worldwide. by the Journal of Gene Medicine. WILEY. Available online: https://a873679.fmphost.com/fmi/webd/GTCT (accessed on 1 October 2023).
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug. Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Herttuala, S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Roubertie, A.; Opladen, T.; Brennenstuhl, H.; Kuseyri Hübschmann, O.; Flint, L.; Willemsen, M.A.; Leuzzi, V.; Cazorla, A.G.; Kurian, M.A.; François-Heude, M.C.; et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency: Requirements for safe application and knowledge-generating follow-up. J. Inherit. Metab. Dis. 2023. [Google Scholar] [CrossRef]
- Herzog, R.W.; VandenDriessche, T.; Ozelo, M.C. First hemophilia B gene therapy approved: More than two decades in the making. Mol. Ther. 2023, 31, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Mahlangu, J.; Kaczmarek, R.; von Drygalski, A.; Shapiro, S.; Chou, S.C.; Ozelo, M.C.; Kenet, G.; Peyvandi, F.; Wang, M.; Madan, B.; et al. Two-Year Outcomes of Valoctocogene Roxaparvovec Therapy for Hemophilia A. N. Engl. J. Med. 2023, 388, 694–705. [Google Scholar] [CrossRef]
- FDA Approves First Gene Therapy for Adults with Severe Hemophilia A. 2023. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-adults-severe-hemophilia (accessed on 1 September 2023).
- Available online: https://www.fda.gov/vaccines-blood-biologics/tissue-tissue-products/elevidys (accessed on 1 September 2023).
- Nonnenmacher, M.; Weber, T. Intracellular transport of recombinant adeno-associated virus vectors. Gene. Ther. 2012, 19, 649–658. [Google Scholar] [CrossRef]
- Riyad, J.M.; Weber, T. Intracellular trafficking of adeno-associated virus (AAV) vectors: Challenges and future directions. Gene Ther. 2021, 28, 683–696. [Google Scholar] [CrossRef]
- Zengel, J.; Carette, J.E. Structural and cellular biology of adeno-associated virus attachment and entry. Adv. Virus Res. 2020, 106, 39–84. [Google Scholar]
- Meyer, N.L.; Chapman, M.S. Adeno-associated virus (AAV) cell entry: Structural insights. Trends Microbiol. 2022, 30, 432–451. [Google Scholar] [CrossRef]
- Kronenberg, S.; Bottcher, B.; von der Lieth, C.W.; Bleker, S.; Kleinschmidt, J.A. A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J. Virol. 2005, 79, 5296–5303. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, S.C.; Samulski, R.J. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J. Virol. 2014, 88, 4132–4144. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Montini, E.; Fuess, S.; Storm, T.A.; Grompe, M.; Kay, M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003, 34, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2020, 39, 47–55. [Google Scholar] [CrossRef]
- Nowrouzi, A.; Penaud-Budloo, M.; Kaeppel, C.; Appelt, U.; Le Guiner, C.; Moullier, P.; von Kalle, C.; Snyder, R.O.; Schmidt, M. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol. Ther. 2012, 20, 1177–1186. [Google Scholar] [CrossRef]
- Summerford, C.; Samulski, R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar] [CrossRef]
- Qing, K.; Mah, C.; Hansen, J.; Zhou, S.; Dwarki, V.; Srivastava, A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999, 5, 71–77. [Google Scholar] [CrossRef]
- Summerford, C.; Bartlett, J.S.; Samulski, R.J. AlphaVbeta5 integrin: A co-receptor for adeno-associated virus type 2 infection. Nat. Med. 1999, 5, 78–82. [Google Scholar] [CrossRef]
- Sanlioglu, S.; Benson, P.K.; Yang, J.; Atkinson, E.M.; Reynolds, T.; Engelhardt, J.F. Endocytosis and nuclear trafficking of adeno-associated virus type 2 are controlled by rac1 and phosphatidylinositol-3 kinase activation. J. Virol. 2000, 74, 9184–9196. [Google Scholar] [CrossRef]
- Asokan, A.; Hamra, J.B.; Govindasamy, L.; Agbandje-McKenna, M.; Samulski, R.J. Adeno-associated virus type 2 contains an integrin alpha5beta1 binding domain essential for viral cell entry. J. Virol. 2006, 80, 8961–8969. [Google Scholar] [CrossRef]
- Kashiwakura, Y.; Tamayose, K.; Iwabuchi, K.; Hirai, Y.; Shimada, T.; Matsumoto, K.; Nakamura, T.; Watanabe, M.; Oshimi, K.; Daida, H. Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J. Virol. 2005, 79, 609–614. [Google Scholar] [CrossRef]
- Akache, B.; Grimm, D.; Pandey, K.; Yant, S.R.; Xu, H.; Kay, M.A. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J. Virol. 2006, 80, 9831–9836. [Google Scholar] [CrossRef]
- Wallen, A.J.; Barker, G.A.; Fein, D.E.; Jing, H.; Diamond, S.L. Enhancers of adeno-associated virus AAV2 transduction via high throughput siRNA screening. Mol. Ther. 2011, 19, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Brown, K.E. Integrin alphaVbeta5 is not involved in adeno-associated virus type 2 (AAV2) infection. Virology 1999, 264, 436–440. [Google Scholar] [CrossRef]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef]
- Chen, C.L.; Jensen, R.L.; Schnepp, B.C.; Connell, M.J.; Shell, R.; Sferra, T.J.; Bartlett, J.S.; Clark, K.R.; Johnson, P.R. Molecular characterization of adeno-associated viruses infecting children. J. Virol. 2005, 79, 14781–14792. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Schmidt, K.; Leder, C.; Muller, O.J.; Wobus, C.E.; Bettinger, K.; Von der Lieth, C.W.; King, J.A.; Kleinschmidt, J.A. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003, 77, 11072–11081. [Google Scholar] [CrossRef]
- Cabanes-Creus, M.; Hallwirth, C.V.; Westhaus, A.; Ng, B.H.; Liao, S.H.Y.; Zhu, E.; Navarro, R.G.; Baltazar, G.; Drouyer, M.; Scott, S.; et al. Restoring the natural tropism of AAV2 vectors for human liver. Sci. Transl. Med. 2020, 12, eaba3312. [Google Scholar] [CrossRef] [PubMed]
- Cabanes-Creus, M.; Westhaus, A.; Navarro, R.G.; Baltazar, G.; Zhu, E.; Amaya, A.K.; Liao, S.H.Y.; Scott, S.; Sallard, E.; Dilworth, K.L.; et al. Attenuation of Heparan Sulfate Proteoglycan Binding Enhances In Vivo Transduction of Human Primary Hepatocytes with AAV2. Mol. Ther. Methods Clin. Dev. 2020, 17, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Govindasamy, L.; Afione, S.; Kaludov, N.; Agbandje-McKenna, M.; Chiorini, J.A. Molecular characterization of the heparin-dependent transduction domain on the capsid of a novel adeno-associated virus isolate, AAV(VR-942). J. Virol. 2008, 82, 8911–8916. [Google Scholar] [CrossRef] [PubMed]
- Lerch, T.F.; Chapman, M.S. Identification of the heparin binding site on adeno-associated virus serotype 3B (AAV-3B). Virology 2012, 423, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Miller, E.; Agbandje-McKenna, M.; Samulski, R.J. Alpha2,3 and alpha2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J. Virol. 2006, 80, 9093–9103. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Pillay, S.; Puschnik, A.S.; Nagamine, C.M.; Cheng, F.; Qiu, J.; Carette, J.E.; Vandenberghe, L.H. An Alternate Route for Adeno-associated Virus (AAV) Entry Independent of AAV Receptor. J. Virol. 2018, 92, e02213-17. [Google Scholar] [CrossRef] [PubMed]
- Kurzeder, C.; Koppold, B.; Sauer, G.; Pabst, S.; Kreienberg, R.; Deissler, H. CD9 promotes adeno-associated virus type 2 infection of mammary carcinoma cells with low cell surface expression of heparan sulphate proteoglycans. Int. J. Mol. Med. 2007, 19, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Steadman, R.A.; Johnson, F.B. Attachment of adeno-associated virus type 3H to fibroblast growth factor receptor 1. Arch. Virol. 2006, 151, 617–623. [Google Scholar] [CrossRef]
- Ling, C.; Lu, Y.; Kalsi, J.K.; Jayandharan, G.R.; Li, B.; Ma, W.; Cheng, B.; Gee, S.W.; McGoogan, K.E.; Govindasamy, L.; et al. Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum. Gene Ther. 2010, 21, 1741–1747. [Google Scholar] [CrossRef]
- Kaludov, N.; Brown, K.E.; Walters, R.W.; Zabner, J.; Chiorini, J.A. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 2001, 75, 6884–6893. [Google Scholar] [CrossRef]
- Di Pasquale, G.; Davidson, B.L.; Stein, C.S.; Martins, I.; Scudiero, D.; Monks, A.; Chiorini, J.A. Identification of PDGFR as a receptor for AAV-5 transduction. Nat. Med. 2003, 9, 1306–1312. [Google Scholar] [CrossRef]
- Weller, M.L.; Amornphimoltham, P.; Schmidt, M.; Wilson, P.A.; Gutkind, J.S.; Chiorini, J.A. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat. Med. 2010, 16, 662–664. [Google Scholar] [CrossRef]
- Shen, S.; Bryant, K.D.; Brown, S.M.; Randell, S.H.; Asokan, A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 2011, 286, 13532–13540. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.L.; Gurda, B.L.; Van Vliet, K.; Agbandje-McKenna, M.; Wilson, J.M. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J. Virol. 2012, 86, 7326–7333. [Google Scholar] [CrossRef] [PubMed]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Yu, J.C.; Hsi, J.; Chipman, P.; Broecker, F.; Fuming, Z.; Linhardt, R.J.; Seeberger, P.H.; Heilbronn, R.; McKenna, R.; et al. Structural Study of Aavrh.10 Receptor and Antibody Interactions. J. Virol. 2021, 95, e0124921. [Google Scholar] [CrossRef] [PubMed]
- Albright, B.H.; Simon, K.E.; Pillai, M.; Devlin, G.W.; Asokan, A. Modulation of Sialic Acid Dependence Influences the Central Nervous System Transduction Profile of Adeno-associated Viruses. J. Virol. 2019, 93, e00332-19. [Google Scholar] [CrossRef]
- Huang, L.Y.; Patel, A.; Ng, R.; Miller, E.B.; Halder, S.; McKenna, R.; Asokan, A.; Agbandje-McKenna, M. Characterization of the Adeno-Associated Virus 1 and 6 Sialic Acid Binding Site. J. Virol. 2016, 90, 5219–5230. [Google Scholar] [CrossRef]
- Rutledge, E.A.; Halbert, C.L.; Russell, D.W. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 1998, 72, 309–319. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 2001, 75, 6615–6624. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Grieger, J.C.; Govindasamy, L.; Agbandje-McKenna, M.; Samulski, R.J. Single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. J. Virol. 2006, 80, 11393–11397. [Google Scholar] [CrossRef]
- Walters, R.W.; Yi, S.M.; Keshavjee, S.; Brown, K.E.; Welsh, M.J.; Chiorini, J.A.; Zabner, J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 2001, 276, 20610–20616. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Fusco, R.M.; Elmore, Z.C.; Asokan, A. Interplay between Furin and Sialoglycans in Modulating Adeno-Associated Viral Cell Entry. J. Virol. 2023, 97, e0009323. [Google Scholar] [CrossRef] [PubMed]
- Bantel-Schaal, U.; zur Hausen, H. Characterization of the DNA of a defective human parvovirus isolated from a genital site. Virology 1984, 134, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Bantel-Schaal, U.; Delius, H.; Schmidt, R.; zur Hausen, H. Human adeno-associated virus type 5 is only distantly related to other known primate helper-dependent parvoviruses. J. Virol. 1999, 73, 939–947. [Google Scholar] [CrossRef]
- Chiorini, J.A.; Kim, F.; Yang, L.; Kotin, R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999, 73, 1309–1319. [Google Scholar] [CrossRef]
- Stutika, C.; Huser, D.; Weger, S.; Rutz, N.; Hessler, M.; Heilbronn, R. Definition of herpes simplex virus helper functions for the replication of adeno-associated virus type 5. J. Gen. Virol. 2015, 96, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Afione, S.; DiMattia, M.A.; Halder, S.; Di Pasquale, G.; Agbandje-McKenna, M.; Chiorini, J.A. Identification and mutagenesis of the adeno-associated virus 5 sialic acid binding region. J. Virol. 2015, 89, 1660–1672. [Google Scholar] [CrossRef]
- Hahm, H.S.; Broecker, F.; Kawasaki, F.; Mietzsch, M.; Heilbronn, R.; Fukuda, M.; Seeberger, P.H. Automated Glycan Assembly of Oligo-N-Acetyllactosamine and Keratan Sulfate Probes to Study Virus-Glycan Interactions. Chem 2017, 2, 114–124. [Google Scholar] [CrossRef]
- Shen, S.; Berry, G.E.; Castellanos Rivera, R.M.; Cheung, R.Y.; Troupes, A.N.; Brown, S.M.; Kafri, T.; Asokan, A. Functional analysis of the putative integrin recognition motif on adeno-associated virus 9. J. Biol. Chem. 2015, 290, 1496–1504. [Google Scholar] [CrossRef]
- Large, E.E.; Chapman, M.S. Adeno-associated virus receptor complexes and implications for adeno-associated virus immune neutralization. Front. Microbiol. 2023, 14, 1116896. [Google Scholar] [CrossRef]
- Dudek, A.M.; Zabaleta, N.; Zinn, E.; Pillay, S.; Zengel, J.; Porter, C.; Franceschini, J.S.; Estelien, R.; Carette, J.E.; Zhou, G.L.; et al. GPR108 Is a Highly Conserved AAV Entry Factor. Mol. Ther. 2020, 28, 367–381. [Google Scholar] [CrossRef]
- Meisen, W.H.; Nejad, Z.B.; Hardy, M.; Zhao, H.; Oliverio, O.; Wang, S.; Hale, C.; Ollmann, M.M.; Collins, P.J. Pooled Screens Identify GPR108 and TM9SF2 as Host Cell Factors Critical for AAV Transduction. Mol. Ther. Methods Clin. Dev. 2020, 17, 601–611. [Google Scholar] [CrossRef]
- Duan, D.; Li, Q.; Kao, A.W.; Yue, Y.; Pessin, J.E.; Engelhardt, J.F. Dynamin is required for recombinant adeno-associated virus type 2 infection. J. Virol. 1999, 73, 10371–10376. [Google Scholar] [CrossRef]
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 2000, 74, 2777–2785. [Google Scholar] [CrossRef]
- Bess, C.D. Analysis of Cellular Factors Involved in Adeno-Associated Virus Type 2 Entry. Ph.D. Thesis, Rockefeller University, New York, NY, USA, 2009. [Google Scholar]
- Lundmark, R.; Doherty, G.J.; Howes, M.T.; Cortese, K.; Vallis, Y.; Parton, R.G.; McMahon, H.T. The GTPase-activating protein GRAF1 regulates the CLIC/GEEC endocytic pathway. Curr. Biol. 2008, 18, 1802–1808. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Sharma, P.; Parton, R.G.; Mayor, S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2002, 2, 411–423. [Google Scholar] [CrossRef]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Braspenning-Wesch, I.; Kartenbeck, J. Adeno-associated virus type 5 exploits two different entry pathways in human embryo fibroblasts. J. Gen. Virol. 2009, 90, 317–322. [Google Scholar] [CrossRef]
- Di Pasquale, G.; Chiorini, J.A. AAV transcytosis through barrier epithelia and endothelium. Mol. Ther. 2006, 13, 506–516. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Hub, B.; Kartenbeck, J. Endocytosis of adeno-associated virus type 5 leads to accumulation of virus particles in the Golgi compartment. J. Virol. 2002, 76, 2340–2349. [Google Scholar] [CrossRef]
- Johnson, J.S.; Gentzsch, M.; Zhang, L.; Ribeiro, C.M.; Kantor, B.; Kafri, T.; Pickles, R.J.; Samulski, R.J. AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis. PLoS Pathog. 2011, 7, e1002053. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, M.E.; Cintrat, J.C.; Gillet, D.; Weber, T. Syntaxin 5-dependent retrograde transport to the trans-Golgi network is required for adeno-associated virus transduction. J. Virol. 2015, 89, 1673–1687. [Google Scholar] [CrossRef]
- Pajusola, K.; Gruchala, M.; Joch, H.; Luscher, T.F.; Yla-Herttuala, S.; Bueler, H. Cell-type-specific characteristics modulate the transduction efficiency of adeno-associated virus type 2 and restrain infection of endothelial cells. J. Virol. 2002, 76, 11530–11540. [Google Scholar] [CrossRef]
- Keiser, N.W.; Yan, Z.; Zhang, Y.; Lei-Butters, D.C.; Engelhardt, J.F. Unique characteristics of AAV1, 2, and 5 viral entry, intracellular trafficking, and nuclear import define transduction efficiency in HeLa cells. Hum. Gene Ther. 2011, 22, 1433–1444. [Google Scholar] [CrossRef]
- Ding, W.; Zhang, L.N.; Yeaman, C.; Engelhardt, J.F. rAAV2 traffics through both the late and the recycling endosomes in a dose-dependent fashion. Mol. Ther. 2006, 13, 671–682. [Google Scholar] [CrossRef]
- Dascher, C.; Matteson, J.; Balch, W.E. Syntaxin 5 regulates endoplasmic reticulum to Golgi transport. J. Biol. Chem. 1994, 269, 29363–29366. [Google Scholar] [CrossRef]
- Meyer, N.L.; Hu, G.; Davulcu, O.; Xie, Q.; Noble, A.J.; Yoshioka, C.; Gingerich, D.S.; Trzynka, A.; David, L.; Stagg, S.M.; et al. Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. eLife 2019, 8, e44707. [Google Scholar] [CrossRef]
- Zhang, R.; Cao, L.; Cui, M.; Sun, Z.; Hu, M.; Zhang, R.; Stuart, W.; Zhao, X.; Yang, Z.; Li, X.; et al. Adeno-associated virus 2 bound to its cellular receptor AAVR. Nat. Microbiol. 2019, 4, 675–682. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, R.; Li, H.; Yin, K.; Ma, X.; Lou, Z. Structural basis for the neurotropic AAV9 and the engineered AAVPHP.eB recognition with cellular receptors. Mol. Ther. Methods Clin. Dev. 2022, 26, 52–60. [Google Scholar] [CrossRef]
- Pillay, S.; Zou, W.; Cheng, F.; Puschnik, A.S.; Meyer, N.L.; Ganaie, S.S.; Deng, X.; Wosen, J.E.; Davulcu, O.; Yan, Z.; et al. Adeno-associated Virus (AAV) Serotypes Have Distinctive Interactions with Domains of the Cellular AAV Receptor. J. Virol. 2017, 91, e00391-17. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, G.; Cao, L.; Sun, Z.; He, Y.; Cui, M.; Sun, Y.; Li, S.; Li, H.; Qin, L. Divergent engagements between adeno-associated viruses with their cellular receptor AAVR. Nat. Commun. 2019, 10, 3760. [Google Scholar] [CrossRef] [PubMed]
- Silveria, M.A.; Large, E.E.; Zane, G.M.; White, T.A.; Chapman, M.S. The Structure of an AAV5-AAVR Complex at 2.5 Å Resolution: Implications for Cellular Entry and Immune Neutralization of AAV Gene Therapy Vectors. Viruses 2020, 12, 1326. [Google Scholar] [CrossRef]
- Douar, A.M.; Poulard, K.; Stockholm, D.; Danos, O. Intracellular trafficking of adeno-associated virus vectors: Routing to the late endosomal compartment and proteasome degradation. J. Virol. 2001, 75, 1824–1833. [Google Scholar] [CrossRef]
- Madigan, V.J.; Berry, G.E.; Tyson, T.O.; Nardone-White, D.; Ark, J.; Elmore, Z.C.; Murlidharan, G.; Vincent, H.A.; Asokan, A. The Golgi Calcium ATPase Pump Plays an Essential Role in Adeno-associated Virus Trafficking and Transduction. J. Virol. 2020, 94, e01604-20. [Google Scholar] [CrossRef] [PubMed]
- Akache, B.; Grimm, D.; Shen, X.; Fuess, S.; Yant, S.R.; Glazer, D.S.; Park, J.; Kay, M.A. A two-hybrid screen identifies cathepsins B and L as uncoating factors for adeno-associated virus 2 and 8. Mol. Ther. 2007, 15, 330–339. [Google Scholar] [CrossRef]
- Stahnke, S.; Lux, K.; Uhrig, S.; Kreppel, F.; Hosel, M.; Coutelle, O.; Ogris, M.; Hallek, M.; Buning, H. Intrinsic phospholipase A2 activity of adeno-associated virus is involved in endosomal escape of incoming particles. Virology 2011, 409, 77–83. [Google Scholar] [CrossRef]
- Zadori, Z.; Szelei, J.; Lacoste, M.C.; Li, Y.; Gariepy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Johnson, J.S.; Samulski, R.J. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J. Virol. 2009, 83, 2632–2644. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, G.; Ried, M.U.; Endress, T.; Buning, H.; Hallek, M.; Brauchle, C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929–1932. [Google Scholar] [CrossRef]
- Thomas, C.E.; Storm, T.A.; Huang, Z.; Kay, M.A. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J. Virol. 2004, 78, 3110–3122. [Google Scholar] [CrossRef] [PubMed]
- Lux, K.; Goerlitz, N.; Schlemminger, S.; Perabo, L.; Goldnau, D.; Endell, J.; Leike, K.; Kofler, D.M.; Finke, S.; Hallek, M.; et al. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 2005, 79, 11776–11787. [Google Scholar] [CrossRef]
- Kelich, J.M.; Ma, J.; Dong, B.; Wang, Q.; Chin, M.; Magura, C.M.; Xiao, W.; Yang, W. Super-resolution imaging of nuclear import of adeno-associated virus in live cells. Mol. Ther. Methods Clin. Dev. 2015, 2, 15047. [Google Scholar] [CrossRef]
- Bevington, J.M.; Needham, P.G.; Verrill, K.C.; Collaco, R.F.; Basrur, V.; Trempe, J.P. Adeno-associated virus interactions with B23/Nucleophosmin: Identification of sub-nucleolar virion regions. Virology 2007, 357, 102–113. [Google Scholar] [CrossRef]
- Qiu, J.; Brown, K.E. A 110-kDa nuclear shuttle protein, nucleolin, specifically binds to adeno-associated virus type 2 (AAV-2) capsid. Virology 1999, 257, 373–382. [Google Scholar] [CrossRef]
- Ros, C.; Baltzer, C.; Mani, B.; Kempf, C. Parvovirus uncoating in vitro reveals a mechanism of DNA release without capsid disassembly and striking differences in encapsidated DNA stability. Virology 2006, 345, 137–147. [Google Scholar] [CrossRef]
- Cotmore, S.F.; D’Abramo, A.M., Jr.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef]
- Sutter, S.O.; Lkharrazi, A.; Schraner, E.M.; Michaelsen, K.; Meier, A.F.; Marx, J.; Vogt, B.; Büning, H.; Fraefel, C. Adeno-associated virus type 2 (AAV2) uncoating is a stepwise process and is linked to structural reorganization of the nucleolus. PLoS Pathog. 2022, 18, e1010187. [Google Scholar] [CrossRef]
- Rossi, A.; Dupaty, L.; Aillot, L.; Zhang, L.; Gallien, C.; Hallek, M.; Odenthal, M.; Adriouch, S.; Salvetti, A.; Buning, H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019, 9, 3631. [Google Scholar] [CrossRef]
- Hsu, H.L.; Brown, A.; Loveland, A.B.; Lotun, A.; Xu, M.; Luo, L.; Xu, G.; Li, J.; Ren, L.; Su, Q.; et al. Structural characterization of a novel human adeno-associated virus capsid with neurotropic properties. Nat. Commun. 2020, 11, 3279. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar] [CrossRef]
- Fisher, K.J.; Gao, G.P.; Weitzman, M.D.; DeMatteo, R.; Burda, J.F.; Wilson, J.M. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 1996, 70, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Afione, S.A.; Conrad, C.K.; Kearns, W.G.; Chunduru, S.; Adams, R.; Reynolds, T.C.; Guggino, W.B.; Cutting, G.R.; Carter, B.J.; Flotte, T.R. In vivo model of adeno-associated virus vector persistence and rescue. J. Virol. 1996, 70, 3235–3241. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Afione, S.A.; Zeitlin, P.L. Adeno-associated virus vector gene expression occurs in nondividing cells in the absence of vector DNA integration. Am. J. Respir. Cell Mol. Biol. 1994, 11, 517–521. [Google Scholar] [CrossRef]
- Schnepp, B.C.; Clark, K.R.; Klemanski, D.L.; Pacak, C.A.; Johnson, P.R. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol. 2003, 77, 3495–3504. [Google Scholar] [CrossRef] [PubMed]
- Schnepp, B.C.; Jensen, R.L.; Chen, C.L.; Johnson, P.R.; Clark, K.R. Characterization of adeno-associated virus genomes isolated from human tissues. J. Virol. 2005, 79, 14793–14803. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Sharma, P.; Dudus, L.; Zhang, Y.; Sanlioglu, S.; Yan, Z.; Yue, Y.; Ye, Y.; Lester, R.; Yang, J.; et al. Formation of adeno-associated virus circular genomes is differentially regulated by adenovirus E4 ORF6 and E2a gene expression. J. Virol. 1999, 73, 161–169. [Google Scholar] [CrossRef]
- Fisher, K.J.; Jooss, K.; Alston, J.; Yang, Y.; Haecker, S.E.; High, K.; Pathak, R.; Raper, S.E.; Wilson, J.M. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med. 1997, 3, 306–312. [Google Scholar] [CrossRef]
- Duan, D.; Sharma, P.; Yang, J.; Yue, Y.; Dudus, L.; Zhang, Y.; Fisher, K.J.; Engelhardt, J.F. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J. Virol. 1998, 72, 8568–8577. [Google Scholar] [CrossRef]
- Vincent-Lacaze, N.; Snyder, R.O.; Gluzman, R.; Bohl, D.; Lagarde, C.; Danos, O. Structure of adeno-associated virus vector DNA following transduction of the skeletal muscle. J. Virol. 1999, 73, 1949–1955. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, W.; Zhang, Y.; Zidon, T.; Ritchie, T.; Engelhardt, J.F. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J. Virol. 1999, 73, 9468–9477. [Google Scholar] [CrossRef]
- Snyder, R.O.; Spratt, S.K.; Lagarde, C.; Bohl, D.; Kaspar, B.; Sloan, B.; Cohen, L.K.; Danos, O. Efficient and stable adeno-associated virus-mediated transduction in the skeletal muscle of adult immunocompetent mice. Hum. Gene Ther. 1997, 8, 1891–1900. [Google Scholar] [CrossRef]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef]
- Brown, M.; Weber, J. Adenoassociated virus has a unique chromatin structure. Can. J. Biochem. 1982, 60, 1001–1005. [Google Scholar] [CrossRef]
- Marcus-Sekura, C.J.; Carter, B.J. Chromatin-like structure of adeno-associated virus DNA in infected cells. J. Virol. 1983, 48, 79–87. [Google Scholar] [CrossRef]
- Penaud-Budloo, M.; Le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Cherel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef]
- Das, A.; Vijayan, M.; Walton, E.M.; Stafford, V.G.; Fiflis, D.N.; Asokan, A. Epigenetic Silencing of Recombinant Adeno-associated Virus Genomes by NP220 and the HUSH Complex. J. Virol. 2022, 96, e0203921. [Google Scholar] [CrossRef]
- Gonzalez-Sandoval, A.; Pekrun, K.; Tsuji, S.; Zhang, F.; Hung, K.L.; Chang, H.Y.; Kay, M.A. The AAV capsid can influence the epigenetic marking of rAAV delivered episomal genomes in a species dependent manner. Nat. Commun. 2023, 14, 2448. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Nepomuceno, I.B.; Messner, A.H.; Moran, M.L.; Batson, E.P.; Dimiceli, S.; Brown, B.W.; Desch, J.K.; Norbash, A.M.; Conrad, C.K.; et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther. 2002, 13, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Reynolds, T.; Moran, M.L.; Moss, R.B.; Wine, J.J.; Flotte, T.R.; Gardner, P. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet 1998, 351, 1702–1703. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Baumann, T.L.; Siffert, J.; Herzog, C.D.; Alterman, R.; Boulis, N.; Turner, D.A.; Stacy, M.; Lang, A.E.; Lozano, A.M.; et al. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 2013, 80, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; VanBrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009, 73, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, G.; Lebranchu, P.; Billaud, F.; Adjali, O.; Schmitt, S.; Bezieau, S.; Pereon, Y.; Valabregue, R.; Ivan, C.; Darmon, C.; et al. Safety and Long-Term Efficacy of AAV4 Gene Therapy in Patients with RPE65 Leber Congenital Amaurosis. Mol. Ther. 2018, 26, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Arabi, F.; Mansouri, V.; Ahmadbeigi, N. Gene therapy clinical trials, where do we go? An overview. Biomed. Pharmacother. 2022, 153, 113324. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 1 August 2023).
- Cehajic-Kapetanovic, J.; Xue, K.; Martinez-Fernandez de la Camara, C.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zerah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef]
- Shen, W.; Liu, S.; Ou, L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: A meta-analysis. Front. Immunol. 2022, 13, 1001263. [Google Scholar] [CrossRef]
- Philippidis, A. After patient death, FDA places hold on Pfizer Duchenne muscular dystrophy gene therapy trial. Hum. Gene Ther. 2022, 33, 111–115. [Google Scholar] [CrossRef]
- Philippidis, A. Novartis confirms deaths of two patients treated with gene therapy Zolgensma. Hum. Gene Ther. 2022, 33, 842–844. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.J.; Nagabhushan Kalburgi, S.; McCown, T.J.; Jude Samulski, R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene. Ther. 2013, 20, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Bell, P.; Vite, C.H.; Louboutin, J.P.; Grant, R.; Bote, E.; Yu, H.; Pukenas, B.; Hurst, R.; Wilson, J.M. Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol. Ther. Methods Clin. Dev. 2014, 1, 14051. [Google Scholar] [CrossRef]
- Szablowski, J.O.; Bar-Zion, A.; Shapiro, M.G. Achieving Spatial and Molecular Specificity with Ultrasound-Targeted Biomolecular Nanotherapeutics. Acc. Chem. Res. 2019, 52, 2427–2434. [Google Scholar] [CrossRef]
- Samaranch, L.; Salegio, E.A.; San Sebastian, W.; Kells, A.P.; Foust, K.D.; Bringas, J.R.; Lamarre, C.; Forsayeth, J.; Kaspar, B.K.; Bankiewicz, K.S. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 2012, 23, 382–389. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide Epidemiology of Neutralizing Antibodies to Adeno-Associated Viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Investig. 2009, 119, 2388–2398. [Google Scholar] [CrossRef]
- Li, C.; He, Y.; Nicolson, S.; Hirsch, M.; Weinberg, M.S.; Zhang, P.; Kafri, T.; Samulski, R.J. Adeno-associated virus capsid antigen presentation is dependent on endosomal escape. J. Clin. Investig. 2013, 123, 1390–1401. [Google Scholar] [CrossRef]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef]
- Duan, D.; Yue, Y.; Yan, Z.; Yang, J.; Engelhardt, J.F. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Investig. 2000, 105, 1573–1587. [Google Scholar] [CrossRef]
- Mingozzi, F.; Maus, M.V.; Hui, D.J.; Sabatino, D.E.; Murphy, S.L.; Rasko, J.E.; Ragni, M.V.; Manno, C.S.; Sommer, J.; Jiang, H.; et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007, 13, 419–422. [Google Scholar] [CrossRef]
- Finn, J.D.; Hui, D.; Downey, H.D.; Dunn, D.; Pien, G.C.; Mingozzi, F.; Zhou, S.; High, K.A. Proteasome inhibitors decrease AAV2 capsid derived peptide epitope presentation on MHC class I following transduction. Mol. Ther. 2010, 18, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.T.; Basner-Tschakarjan, E.; Markusic, D.M.; Finn, J.D.; Hinderer, C.; Zhou, S.; Ostrov, D.A.; Srivastava, A.; Ertl, H.C.; Terhorst, C.; et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood 2013, 121, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Guenther, C.M.; Suh, J. Adeno-Associated Virus (AAV) Vectors: Rational Design Strategies for Capsid Engineering. Curr. Opin. Biomed. Eng. 2018, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Bartel, M.A.; Weinstein, J.R.; Schaffer, D.V. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene. Ther. 2012, 19, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Enoki, T.; Kawano, Y.; Veraz, M.; Nakai, H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat. Commun. 2014, 5, 3075. [Google Scholar] [CrossRef] [PubMed]
- Boucas, J.; Lux, K.; Huber, A.; Schievenbusch, S.; von Freyend, M.J.; Perabo, L.; Quadt-Humme, S.; Odenthal, M.; Hallek, M.; Buning, H. Engineering adeno-associated virus serotype 2-based targeting vectors using a new insertion site-position 453-and single point mutations. J. Gene Med. 2009, 11, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Girod, A.; Ried, M.; Wobus, C.; Lahm, H.; Leike, K.; Kleinschmidt, J.; Deleage, G.; Hallek, M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 1999, 5, 1052–1056. [Google Scholar] [CrossRef]
- Rabinowitz, J.E.; Xiao, W.; Samulski, R.J. Insertional mutagenesis of AAV2 capsid and the production of recombinant virus. Virology 1999, 265, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Marsic, D.; Govindasamy, L.; Currlin, S.; Markusic, D.M.; Tseng, Y.S.; Herzog, R.W.; Agbandje-McKenna, M.; Zolotukhin, S. Vector design Tour de Force: Integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol. Ther. 2014, 22, 1900–1909. [Google Scholar] [CrossRef]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013, 5, 189ra176. [Google Scholar] [CrossRef]
- Grimm, D.; Lee, J.S.; Wang, L.; Desai, T.; Akache, B.; Storm, T.A.; Kay, M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008, 82, 5887–5911. [Google Scholar] [CrossRef]
- Perabo, L.; Endell, J.; King, S.; Lux, K.; Goldnau, D.; Hallek, M.; Buning, H. Combinatorial engineering of a gene therapy vector: Directed evolution of adeno-associated virus. J. Gene Med. 2006, 8, 155–162. [Google Scholar] [CrossRef]
- Koerber, J.T.; Jang, J.H.; Schaffer, D.V. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 2008, 16, 1703–1709. [Google Scholar] [CrossRef]
- Kotin, R.; Hou, J.; McLaughlin, J. Chimeric Capsids. U.S. Patent 2020/0149068 A1, 14 May 2020. [Google Scholar]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sanchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Merkel, S.F.; Andrews, A.M.; Lutton, E.M.; Mu, D.; Hudry, E.; Hyman, B.T.; Maguire, C.A.; Ramirez, S.H. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J. Neurochem. 2017, 140, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef]
- Batista, A.R.; King, O.D.; Reardon, C.P.; Davis, C.; Shankaracharya; Philip, V.; Gray-Edwards, H.; Aronin, N.; Lutz, C.; Landers, J.; et al. Ly6a Differential Expression in Blood-Brain Barrier Is Responsible for Strain Specific Central Nervous System Transduction Profile of AAV-PHP.B. Hum. Gene Ther. 2020, 31, 90–102. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Tanaka, M.; Hakoda, S.; Masuda, T.; Miyata, R.; Konno, A.; Hirai, H. Neurotropic Properties of AAV-PHP.B Are Shared among Diverse Inbred Strains of Mice. Mol. Ther. 2019, 27, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Challis, R.C.; Ravindra Kumar, S.; Chan, K.Y.; Challis, C.; Beadle, K.; Jang, M.J.; Kim, H.M.; Rajendran, P.S.; Tompkins, J.D.; Shivkumar, K.; et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc. 2019, 14, 379–414. [Google Scholar] [CrossRef]
- Jackson, K.L.; Dayton, R.D.; Deverman, B.E.; Klein, R.L. Better Targeting, Better Efficiency for Wide-Scale Neuronal Transduction with the Synapsin Promoter and AAV-PHP.B. Front. Mol. Neurosci. 2016, 9, 116. [Google Scholar]
- Ravindra Kumar, S.; Miles, T.F.; Chen, X.; Brown, D.; Dobreva, T.; Huang, Q.; Ding, X.; Luo, Y.; Einarsson, P.H.; Greenbaum, A.; et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods 2020, 17, 541–550. [Google Scholar] [CrossRef]
- Goertsen, D.; Flytzanis, N.C.; Goeden, N.; Chuapoco, M.R.; Cummins, A.; Chen, Y.; Fan, Y.; Zhang, Q.; Sharma, J.; Duan, Y.; et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022, 25, 106–115. [Google Scholar] [CrossRef]
- Chuapoco, M.R.; Flytzanis, N.C.; Goeden, N.; Christopher Octeau, J.; Roxas, K.M.; Chan, K.Y.; Scherrer, J.; Winchester, J.; Blackburn, R.J.; Campos, L.J.; et al. Adeno-associated viral vectors for functional intravenous gene transfer throughout the non-human primate brain. Nat. Nanotechnol. 2023, 18, 1241–1251. [Google Scholar] [CrossRef]
- Brown, D.; Altermatt, M.; Dobreva, T.; Chen, S.; Wang, A.; Thomson, M.; Gradinaru, V. Deep Parallel Characterization of AAV Tropism and AAV-Mediated Transcriptional Changes via Single-Cell RNA Sequencing. Front. Immunol. 2021, 12, 730825. [Google Scholar] [CrossRef]
- Jang, M.J.; Coughlin, G.M.; Jackson, C.R.; Chen, X.; Chuapoco, M.R.; Vendemiatti, J.L.; Wang, A.Z.; Gradinaru, V. Spatial transcriptomics for profiling the tropism of viral vectors in tissues. Nat. Biotechnol. 2023, 41, 1272–1286. [Google Scholar] [CrossRef]
- Wang, S.K.; Lapan, S.W.; Hong, C.M.; Krause, T.B.; Cepko, C.L. In Situ Detection of Adeno-associated Viral Vector Genomes with SABER-FISH. Mol. Ther. Methods Clin. Dev. 2020, 19, 376–386. [Google Scholar] [CrossRef]
- Yang, B.; Treweek, J.B.; Kulkarni, R.P.; Deverman, B.E.; Chen, C.K.; Lubeck, E.; Shah, S.; Cai, L.; Gradinaru, V. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell 2014, 158, 945–958. [Google Scholar] [CrossRef]
- Terashima, T.; Ogawa, N.; Nakae, Y.; Sato, T.; Katagi, M.; Okano, J.; Maegawa, H.; Kojima, H. Gene Therapy for Neuropathic Pain through siRNA-IRF5 Gene Delivery with Homing Peptides to Microglia. Mol. Ther. Nucleic Acids 2018, 11, 203–215. [Google Scholar] [CrossRef]
- Chen, X.; Wolfe, D.A.; Bindu, D.S.; Zhang, M.; Taskin, N.; Goertsen, D.; Shay, T.F.; Sullivan, E.; Huang, S.F.; Kumar, S.R.; et al. Functional gene delivery to and across brain vasculature of systemic AAVs with endothelial-specific tropism in rodents and broad tropism in primates. bioRxiv 2023. [Google Scholar] [CrossRef]
- Pietersz, K.L.; Plessis, F.D.; Pouw, S.M.; Liefhebber, J.M.; van Deventer, S.J.; Martens, G.J.M.; Konstantinova, P.S.; Blits, B. PhP.B Enhanced Adeno-Associated Virus Mediated-Expression Following Systemic Delivery or Direct Brain Administration. Front. Bioeng. Biotechnol. 2021, 9, 679483. [Google Scholar] [CrossRef]
- Martino, R.A.; Fluck, E.C., 3rd; Murphy, J.; Wang, Q.; Hoff, H.; Pumroy, R.A.; Lee, C.Y.; Sims, J.J.; Roy, S.; Moiseenkova-Bell, V.Y.; et al. Context-Specific Function of the Engineered Peptide Domain of PHP.B. J. Virol. 2021, 95, e0116421. [Google Scholar] [CrossRef]
- Pulicherla, N.; Shen, S.; Yadav, S.; Debbink, K.; Govindasamy, L.; Agbandje-McKenna, M.; Asokan, A. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol. Ther. 2011, 19, 1070–1078. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Harris, A.F.; Cabral, D.J.; Keeler, A.M.; Sapp, E.; Ferreira, J.S.; Gray-Edwards, H.L.; Johnson, J.A.; Johnson, A.K.; Su, Q.; et al. Widespread Central Nervous System Gene Transfer and Silencing After Systemic Delivery of Novel AAV-AS Vector. Mol. Ther. 2016, 24, 726–735. [Google Scholar] [CrossRef]
- Jang, J.H.; Koerber, J.T.; Kim, J.S.; Asuri, P.; Vazin, T.; Bartel, M.; Keung, A.; Kwon, I.; Park, K.I.; Schaffer, D.V. An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells. Mol. Ther. 2011, 19, 667–675. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Vazin, T.; Schaffer, D.V. Enhanced selective gene delivery to neural stem cells in vivo by an adeno-associated viral variant. Development 2015, 142, 1885–1892. [Google Scholar] [CrossRef]
- Shay, T.F.; Sullivan, E.E.; Ding, X.; Chen, X.; Ravindra Kumar, S.; Goertsen, D.; Brown, D.; Crosby, A.; Vielmetter, J.; Borsos, M.; et al. Primate-conserved carbonic anhydrase IV and murine-restricted LY6C1 enable blood-brain barrier crossing by engineered viral vectors. Sci. Adv. 2023, 9, eadg6618. [Google Scholar] [CrossRef]
- Eid, F.-E.; Chen, A.T.; Chan, K.Y.; Huang, Q.; Zheng, Q.; Tobey, I.G.; Pacouret, S.; Brauer, P.P.; Keyes, C.; Powell, M.; et al. Systematic multi-trait AAV capsid engineering for efficient gene delivery. bioRxiv 2022. [Google Scholar] [CrossRef]
- Krolak, T.; Chan, K.Y.; Kaplan, L.; Huang, Q.; Wu, J.; Zheng, Q.; Kozareva, V.; Beddow, T.; Tobey, I.G.; Pacouret, S.; et al. A High-Efficiency AAV for Endothelial Cell Transduction Throughout the Central Nervous System. Nat. Cardiovasc. Res. 2022, 1, 389–400. [Google Scholar] [CrossRef]
- Chuapoco, M.R.; Flytzanis, N.C.; Goeden, N.; Octeau, J.C.; Roxas, K.M.; Chan, K.Y.; Scherrer, J.; Winchester, J.; Blackburn, R.J.; Campos, L.J.; et al. Intravenous functional gene transfer throughout the brain of non-human primates using AAV. Res. Sq. 2023. preprint. [Google Scholar]
- Hanlon, K.S.; Meltzer, J.C.; Buzhdygan, T.; Cheng, M.J.; Sena-Esteves, M.; Bennett, R.E.; Sullivan, T.P.; Razmpour, R.; Gong, Y.; Ng, C.; et al. Selection of an Efficient AAV Vector for Robust CNS Transgene Expression. Mol. Ther. Methods Clin. Dev. 2019, 15, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.; Gong, Y.; Kim, J.C.; Hanlon, K.S.; Nammour, J.; Hieber, K.; Eichler, F.; Cheng, M.; Stemmer-Rachamimov, A.; Stankovic, K.M.; et al. The AAV9 Variant Capsid AAV-F Mediates Widespread Transgene Expression in Nonhuman Primate Spinal Cord After Intrathecal Administration. Hum. Gene Ther. 2022, 33, 61–75. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Wang, W.; Child, M.A.; Ren, X.Q.; Huang, C.; Ren, A.Z.; Tocci, J.; Chen, Q.; Bittner, K.; Tyson, K.; et al. Rapid evolution of blood-brain-barrier-penetrating AAV capsids by RNA-driven biopanning. Mol. Ther. Methods Clin. Dev. 2021, 20, 366–378. [Google Scholar] [CrossRef]
- Giannelli, S.G.; Luoni, M.; Bellinazzi, B.; Iannielli, A.; Middeldorp, J.; Philippens, I.; Körbelin, J.; Broccoli, V. New AAV9 engineered variants with enhanced neurotropism and reduced liver off-targeting in mice and marmosets. bioRxiv 2023. [Google Scholar] [CrossRef]
- Korbelin, J.; Dogbevia, G.; Michelfelder, S.; Ridder, D.A.; Hunger, A.; Wenzel, J.; Seismann, H.; Lampe, M.; Bannach, J.; Pasparakis, M.; et al. A brain microvasculature endothelial cell-specific viral vector with the potential to treat neurovascular and neurological diseases. EMBO Mol. Med. 2016, 8, 609–625. [Google Scholar] [CrossRef]
- Tervo, D.G.; Hwang, B.Y.; Viswanathan, S.; Gaj, T.; Lavzin, M.; Ritola, K.D.; Lindo, S.; Michael, S.; Kuleshova, E.; Ojala, D.; et al. A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 2016, 92, 372–382. [Google Scholar] [CrossRef]
- Chen, Z.; Fan, G.; Li, A.; Yuan, J.; Xu, T. rAAV2-Retro Enables Extensive and High-Efficient Transduction of Lower Motor Neurons following Intramuscular Injection. Mol. Ther. Methods Clin. Dev. 2020, 17, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ravindra Kumar, S.; Adams, C.D.; Yang, D.; Wang, T.; Wolfe, D.A.; Arokiaraj, C.M.; Ngo, V.; Campos, L.J.; Griffiths, J.A.; et al. Engineered AAVs for non-invasive gene delivery to rodent and non-human primate nervous systems. Neuron 2022, 110, 2242–2257.e6. [Google Scholar] [CrossRef]
- Ozturk, B.E.; Johnson, M.E.; Kleyman, M.; Turunc, S.; He, J.; Jabalameli, S.; Xi, Z.; Visel, M.; Dufour, V.L.; Iwabe, S.; et al. scAAVengr, a transcriptome-based pipeline for quantitative ranking of engineered AAVs with single-cell resolution. eLife 2021, 10, e64175. [Google Scholar] [CrossRef]
- Ivanchenko, M.V.; Hanlon, K.S.; Hathaway, D.M.; Klein, A.J.; Peters, C.W.; Li, Y.; Tamvakologos, P.I.; Nammour, J.; Maguire, C.A.; Corey, D.P. AAV-S: A versatile capsid variant for transduction of mouse and primate inner ear. Mol. Ther. Methods Clin. Dev. 2021, 21, 382–398. [Google Scholar] [CrossRef]
- Gonzalez, T.J.; Simon, K.E.; Blondel, L.O.; Fanous, M.M.; Roger, A.L.; Maysonet, M.S.; Devlin, G.W.; Smith, T.J.; Oh, D.K.; Havlik, L.P.; et al. Cross-species evolution of a highly potent AAV variant for therapeutic gene transfer and genome editing. Nat. Commun. 2022, 13, 5947. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S. High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020, 38, 910. [Google Scholar]
- Mullard, A. Gene therapy community grapples with toxicity issues, as pipeline matures. Nat. Rev. Drug. Discov. 2021, 20, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, J.; Weis, S.; Sippel, J.; Tulalamba, W.; Remes, A.; El Andari, J.; Herrmann, A.K.; Pham, Q.H.; Borowski, C.; Hille, S.; et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020, 11, 5432. [Google Scholar] [CrossRef]
- Borner, K.; Kienle, E.; Huang, L.Y.; Weinmann, J.; Sacher, A.; Bayer, P.; Stullein, C.; Fakhiri, J.; Zimmermann, L.; Westhaus, A.; et al. Pre-arrayed Pan-AAV Peptide Display Libraries for Rapid Single-Round Screening. Mol. Ther. 2020, 28, 1016–1032. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938 e4922. [Google Scholar] [CrossRef]
- Work, L.M.; Buning, H.; Hunt, E.; Nicklin, S.A.; Denby, L.; Britton, N.; Leike, K.; Odenthal, M.; Drebber, U.; Hallek, M.; et al. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol. Ther. 2006, 13, 683–693. [Google Scholar] [CrossRef]
- Korbelin, J.; Sieber, T.; Michelfelder, S.; Lunding, L.; Spies, E.; Hunger, A.; Alawi, M.; Rapti, K.; Indenbirken, D.; Muller, O.J.; et al. Pulmonary Targeting of Adeno-associated Viral Vectors by Next-generation Sequencing-guided Screening of Random Capsid Displayed Peptide Libraries. Mol. Ther. 2016, 24, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Wang, X.; Yan, R.; Hu, W.; Li, A.; Wang, S.; Li, H. Single point mutation in adeno-associated viral vectors-DJ capsid leads to improvement for gene delivery in vivo. BMC Biotechnol. 2016, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Excoffon, K.J.; Koerber, J.T.; Dickey, D.D.; Murtha, M.; Keshavjee, S.; Kaspar, B.K.; Zabner, J.; Schaffer, D.V. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3865–3870. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, J.; Drouin, L.M.; Agbandje-McKenna, M.; Chen, C.; Qiao, C.; Pu, D.; Hu, X.; Wang, D.Z.; Li, J.; et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc. Natl. Acad. Sci. USA 2009, 106, 3946–3951. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.S.; Sun, S.; Santiago-Ortiz, J.L.; Shapiro, M.G.; Romero, P.A.; Schaffer, D.V. In Vivo Selection of a Computationally Designed SCHEMA AAV Library Yields a Novel Variant for Infection of Adult Neural Stem Cells in the SVZ. Mol. Ther. 2018, 26, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Fitzpatrick, Z.; Harris, A.F.; Maitland, S.A.; Ferreira, J.S.; Zhang, Y.; Ma, S.; Sharma, R.B.; Gray-Edwards, H.L.; Johnson, J.A.; et al. In Vivo Selection Yields AAV-B1 Capsid for Central Nervous System and Muscle Gene Therapy. Mol. Ther. 2016, 24, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef]
- Pekrun, K.; De Alencastro, G.; Luo, Q.J.; Liu, J.; Kim, Y.; Nygaard, S.; Galivo, F.; Zhang, F.; Song, R.; Tiffany, M.R.; et al. Using a barcoded AAV capsid library to select for clinically relevant gene therapy vectors. JCI Insight 2019, 4, e131610. [Google Scholar] [CrossRef]
- Shen, S.; Troupes, A.N.; Pulicherla, N.; Asokan, A. Multiple roles for sialylated glycans in determining the cardiopulmonary tropism of adeno-associated virus 4. J. Virol. 2013, 87, 13206–13213. [Google Scholar] [CrossRef]
- Murlidharan, G.; Corriher, T.; Ghashghaei, H.T.; Asokan, A. Unique glycan signatures regulate adeno-associated virus tropism in the developing brain. J. Virol. 2015, 89, 3976–3987. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef]
- Asokan, A.; Conway, J.C.; Phillips, J.L.; Li, C.; Hegge, J.; Sinnott, R.; Yadav, S.; DiPrimio, N.; Nam, H.J.; Agbandje-McKenna, M.; et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat. Biotechnol. 2010, 28, 79–82. [Google Scholar] [CrossRef]
- Shen, S.; Horowitz, E.D.; Troupes, A.N.; Brown, S.M.; Pulicherla, N.; Samulski, R.J.; Agbandje-McKenna, M.; Asokan, A. Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J. Biol. Chem. 2013, 288, 28814–28823. [Google Scholar] [CrossRef]
- Zhong, L.; Li, B.; Mah, C.S.; Govindasamy, L.; Agbandje-McKenna, M.; Cooper, M.; Herzog, R.W.; Zolotukhin, I.; Warrington, K.H., Jr.; Weigel-Van Aken, K.A.; et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA 2008, 105, 7827–7832. [Google Scholar] [CrossRef]
- Petrs-Silva, H.; Dinculescu, A.; Li, Q.; Min, S.H.; Chiodo, V.; Pang, J.J.; Zhong, L.; Zolotukhin, S.; Srivastava, A.; Lewin, A.S.; et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol. Ther. 2009, 17, 463–471. [Google Scholar] [CrossRef]
- Albright, B.H.; Storey, C.M.; Murlidharan, G.; Castellanos Rivera, R.M.; Berry, G.E.; Madigan, V.J.; Asokan, A. Mapping the Structural Determinants Required for AAVrh.10 Transport across the Blood-Brain Barrier. Mol. Ther. 2018, 26, 510–523. [Google Scholar] [CrossRef]
- Yang, B.; Li, S.; Wang, H.; Guo, Y.; Gessler, D.J.; Cao, C.; Su, Q.; Kramer, J.; Zhong, L.; Ahmed, S.S.; et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 2014, 22, 1299–1309. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Mu, X.; Ahmed, S.S.; Su, Q.; He, R.; Wang, H.; Mueller, C.; Sena-Esteves, M.; Brown, R.; et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 2011, 19, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, A.T.; Chan, K.Y.; Sorensen, H.; Barry, A.J.; Azari, B.; Beddow, T.; Zheng, Q.; Zhao, B.; Tobey, I.G.; et al. Targeting AAV vectors to the CNS via de novo engineered capsid-receptor interactions. bioRxiv 2022. [Google Scholar] [CrossRef]
- Huang, Q.; Chan, K.Y.; Lou, S.; Keyes, C.; Wu, J.; Botticello-Romero, N.R.; Zheng, Q.; Johnston, J.; Mills, A.; Brauer, P.P.; et al. An AAV capsid reprogrammed to bind human Transferrin Receptor mediates brain-wide gene delivery. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tan, F.; Chu, C.; Qi, J.; Li, W.; You, D.; Li, K.; Chen, X.; Zhao, W.; Cheng, C.; Liu, X.; et al. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 2019, 10, 3733. [Google Scholar] [CrossRef]
- Davidsson, M.; Wang, G.; Aldrin-Kirk, P.; Cardoso, T.; Nolbrant, S.; Hartnor, M.; Mudannayake, J.; Parmar, M.; Bjorklund, T. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 27053–27062. [Google Scholar] [CrossRef] [PubMed]
- Zinn, E.; Pacouret, S.; Khaychuk, V.; Turunen, H.T.; Carvalho, L.S.; Andres-Mateos, E.; Shah, S.; Shelke, R.; Maurer, A.C.; Plovie, E.; et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015, 12, 1056–1068. [Google Scholar] [CrossRef]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017, 35, 280–284. [Google Scholar] [CrossRef]
- Zinn, E.; Unzu, C.; Schmit, P.F.; Turunen, H.T.; Zabaleta, N.; Sanmiguel, J.; Fieldsend, A.; Bhatt, U.; Diop, C.; Merkel, E.; et al. Ancestral library identifies conserved reprogrammable liver motif on AAV capsid. Cell Rep. Med. 2022, 3, 100803. [Google Scholar] [CrossRef]
- Wong, A.S.; Choi, G.C.; Cheng, A.A.; Purcell, O.; Lu, T.K. Massively parallel high-order combinatorial genetics in human cells. Nat. Biotechnol. 2015, 33, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.; Choi, G.C.; Cui, C.H.; Pregernig, G.; Milani, P.; Adam, M.; Perli, S.D.; Kazer, S.W.; Gaillard, A.; Hermann, M.; et al. Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc. Natl. Acad. Sci. USA 2016, 113, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Cabanes-Creus, M.; Navarro, R.G.; Liao, S.H.Y.; Baltazar, G.; Drouyer, M.; Zhu, E.; Scott, S.; Luong, C.; Wilson, L.O.W.; Alexander, I.E.; et al. Single amino acid insertion allows functional transduction of murine hepatocytes with human liver tropic AAV capsids. Mol. Ther. Methods Clin. Dev. 2021, 21, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE 2019, 14, e0225206. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Yuan, Y.; Clark, P.M.; Wang, Q.; Martino, R.A.; Sims, J.J.; Bell, P.; Raymond, A.; Stanford, W.L.; Wilson, J.M. The GPI-Linked Protein LY6A Drives AAV-PHP.B Transport across the Blood-Brain Barrier. Mol. Ther. 2019, 27, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, G. Emerging Role of Lymphocyte Antigen-6 Family of Genes in Cancer and Immune Cells. Front. Immunol. 2019, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Stanford, W.L. Concise review: Stem cell antigen-1: Expression, function, and enigma. Stem Cells 2007, 25, 1339–1347. [Google Scholar] [CrossRef]
- Jang, S.; Shen, H.K.; Ding, X.; Miles, T.F.; Gradinaru, V. Structural basis of receptor usage by the engineered capsid AAV-PHP.eB. Mol. Ther. Methods Clin. Dev. 2022, 26, 343–354. [Google Scholar] [CrossRef]
- Freeth, J.; Soden, J. New Advances in Cell Microarray Technology to Expand Applications in Target Deconvolution and Off-Target Screening. SLAS Discov. 2020, 25, 223–230. [Google Scholar] [CrossRef]
- Shay, T.F.; Jang, S.; Chen, X.; Walker, B.; Tebbutt, C.; Wolfe, D.A.; Brittain, T.J.; Arokiaraj, C.M.; Sullivan, E.E.; Ding, X.; et al. Human cell surface-AAV interactomes identify LRP6 as blood-brain-barrier transcytosis receptor and immune cytokine IL3 as AAV9 binder. bioRxiv 2024. [Google Scholar] [CrossRef]
- Spangrude, G.J.; Brooks, D.M. Mouse strain variability in the expression of the hematopoietic stem cell antigen Ly-6A/E by bone marrow cells. Blood 1993, 82, 3327–3332. [Google Scholar] [CrossRef]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Loughner, C.L.; Bruford, E.A.; McAndrews, M.S.; Delp, E.E.; Swamynathan, S.; Swamynathan, S.K. Organization, evolution and functions of the human and mouse Ly6/uPAR family genes. Hum. Genom. 2016, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Liguore, W.A.; Domire, J.S.; Button, D.; Wang, Y.; Dufour, B.D.; Srinivasan, S.; McBride, J.L. AAV-PHP.B Administration Results in a Differential Pattern of CNS Biodistribution in Non-human Primates Compared with Mice. Mol. Ther. 2019, 27, 2018–2037. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Konno, A.; Mochizuki, R.; Shinohara, Y.; Nitta, K.; Okada, Y.; Hirai, H. Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 2018, 665, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Shmerling, M.; Chalik, M.; Smorodinsky, N.I.; Meeker, A.; Roy, S.; Sagi-Assif, O.; Meshel, T.; Danilevsky, A.; Shomron, N.; Levinger, S.; et al. LY6S, a New IFN-Inducible Human Member of the Ly6a Subfamily Expressed by Spleen Cells and Associated with Inflammation and Viral Resistance. Immunohorizons 2022, 6, 253–272. [Google Scholar] [CrossRef]
- Lee, P.Y.; Wang, J.X.; Parisini, E.; Dascher, C.C.; Nigrovic, P.A. Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 2013, 94, 585–594. [Google Scholar] [CrossRef]
- Schaffenrath, J.; Huang, S.F.; Wyss, T.; Delorenzi, M.; Keller, A. Characterization of the blood-brain barrier in genetically diverse laboratory mouse strains. Fluids Barriers CNS 2021, 18, 34. [Google Scholar] [CrossRef]
- Ghandour, M.S.; Langley, O.K.; Zhu, X.L.; Waheed, A.; Sly, W.S. Carbonic anhydrase IV on brain capillary endothelial cells: A marker associated with the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1992, 89, 6823–6827. [Google Scholar] [CrossRef]
- Bonfanti, L.; Seki, T. The PSA-NCAM-Positive “Immature” Neurons: An Old Discovery Providing New Vistas on Brain Structural Plasticity. Cells 2021, 10, 2542. [Google Scholar] [CrossRef]
- Davidson, G. LRPs in WNT Signalling. Handb. Exp. Pharmacol. 2021, 269, 45–73. [Google Scholar] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, J.; Liu, Y. LRP5 and LRP6 in Wnt Signaling: Similarity and Divergence. Front. Cell Dev. Biol. 2021, 9, 670960. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Kigoshi, T.; Murasaki, S.; Arai, F.; Shimada, K.; Seki, N.; Kim, Y.G.; Hase, K.; Ohno, H.; Kawano, K.; et al. Pancreatic glycoprotein 2 is a first line of defense for mucosal protection in intestinal inflammation. Nat. Commun. 2021, 12, 1067. [Google Scholar] [CrossRef]
- Sjostedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Liu, A.P.; Aguet, F.; Danuser, G.; Schmid, S.L. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol. 2010, 191, 1381–1393. [Google Scholar] [CrossRef]
- Navaroli, D.M.; Bellve, K.D.; Standley, C.; Lifshitz, L.M.; Cardia, J.; Lambright, D.; Leonard, D.; Fogarty, K.E.; Corvera, S. Rabenosyn-5 defines the fate of the transferrin receptor following clathrin-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2012, 109, E471–E480. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Delaney, C.E.; Brunette, E.; Baumann, E.; Farrington, G.K.; Sisk, W.; Eldredge, J.; Ding, W.; Tremblay, T.L.; Stanimirovic, D.B. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood brain barrier. J. Cereb. Blood Flow Metab. 2018, 38, 727–740. [Google Scholar] [CrossRef]
- Alam, P.; Holst, M.R.; Lauritsen, L.; Nielsen, J.; Nielsen, S.S.E.; Jensen, P.H.; Brewer, J.R.; Otzen, D.E.; Nielsen, M.S. Polarized alpha-synuclein trafficking and transcytosis across brain endothelial cells via Rab7-decorated carriers. Fluids Barriers CNS 2022, 19, 37. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Naumer, M.; Ying, Y.; Michelfelder, S.; Reuter, A.; Trepel, M.; Muller, O.J.; Kleinschmidt, J.A. Development and validation of novel AAV2 random libraries displaying peptides of diverse lengths and at diverse capsid positions. Hum. Gene Ther. 2012, 23, 492–507. [Google Scholar] [CrossRef]
- Varadi, K.; Michelfelder, S.; Korff, T.; Hecker, M.; Trepel, M.; Katus, H.A.; Kleinschmidt, J.A.; Muller, O.J. Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene. Ther. 2012, 19, 800–809. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Isakova, A.; Fehlmann, T.; Keller, A.; Quake, S.R. A mouse tissue atlas of small noncoding RNA. Proc. Natl. Acad. Sci. USA 2020, 117, 25634–25645. [Google Scholar] [CrossRef]
- Hordeaux, J.; Buza, E.L.; Dyer, C.; Goode, T.; Mitchell, T.W.; Richman, L.; Denton, N.; Hinderer, C.; Katz, N.; Schmid, R.; et al. Adeno-Associated Virus-Induced Dorsal Root Ganglion Pathology. Hum. Gene Ther. 2020, 31, 808–818. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef]
- Van Alstyne, M.; Tattoli, I.; Delestree, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. 2021, 24, 930–940. [Google Scholar] [CrossRef]
- Johnston, S.; Parylak, S.L.; Kim, S.; Mac, N.; Lim, C.; Gallina, I.; Bloyd, C.; Newberry, A.; Saavedra, C.D.; Novak, O.; et al. AAV ablates neurogenesis in the adult murine hippocampus. eLife 2021, 10, e59291. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022, 13, 975803. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Schaffer, D.V. Designer gene delivery vectors: Molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm. Res. 2008, 25, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Bertin, B.; Veron, P.; Leborgne, C.; Deschamps, J.Y.; Moullec, S.; Fromes, Y.; Collaud, F.; Boutin, S.; Latournerie, V.; van Wittenberghe, L.; et al. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci. Rep. 2020, 10, 864. [Google Scholar] [CrossRef]
- Orlowski, A.; Weber, T. Selective Anti-AAV Antibody Depletion by Hemapheresis and Immunoadsorption. Methods Mol. Biol. 2022, 2573, 235–248. [Google Scholar]
- Grover, R.K.; Zhu, X.; Nieusma, T.; Jones, T.; Boreo, I.; MacLeod, A.S.; Mark, A.; Niessen, S.; Kim, H.J.; Kong, L.; et al. A structurally distinct human mycoplasma protein that generically blocks antigen-antibody union. Science 2014, 343, 656–661. [Google Scholar] [CrossRef]
- Leborgne, C.; Barbon, E.; Alexander, J.M.; Hanby, H.; Delignat, S.; Cohen, D.M.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J.; et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef]
- Bobo, T.A.; Samowitz, P.N.; Robinson, M.I.; Montes, L.I.; Forsberg, L.J.; Feng, R.; Nicely, N.I.; Fu, H. IgG-cleavage protein allows therapeutic AAV gene delivery in passively immunized MPS IIIA mice. Gene. Ther. 2023, 30, 377–385. [Google Scholar] [CrossRef]
- Ros-Ganan, I.; Hommel, M.; Trigueros-Motos, L.; Tamarit, B.; Rodriguez-Garcia, E.; Salas, D.; Perez, G.; Douar, A.; Combal, J.P.; Benichou, B.; et al. Optimising the IgG-degrading enzyme treatment regimen for enhanced adeno-associated virus transduction in the presence of neutralising antibodies. Clin. Transl. Immunol. 2022, 11, e1375. [Google Scholar] [CrossRef] [PubMed]
- Elmore, Z.C.; Oh, D.K.; Simon, K.E.; Fanous, M.M.; Asokan, A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight 2020, 5, e139881. [Google Scholar] [CrossRef]
- Lopez-Gordo, E.; Orlowski, A.; Wang, A.; Weinberg, A.; Sahoo, S.; Weber, T. Hydroxylation of N-acetylneuraminic Acid Influences the in vivo Tropism of N-linked Sialic Acid-Binding Adeno-Associated Viruses AAV1, AAV5, and AAV6. Front. Med. 2021, 8, 732095. [Google Scholar] [CrossRef]
- Maguire, C.A.; Crommentuijn, M.H.; Mu, D.; Hudry, E.; Serrano-Pozo, A.; Hyman, B.T.; Tannous, B.A. Mouse gender influences brain transduction by intravascularly administered AAV9. Mol. Ther. 2013, 21, 1470–1471. [Google Scholar] [CrossRef]
- Rumachik, N.G.; Malaker, S.A.; Poweleit, N.; Maynard, L.H.; Adams, C.M.; Leib, R.D.; Cirolia, G.; Thomas, D.; Stamnes, S.; Holt, K.; et al. Methods Matter: Standard Production Platforms for Recombinant AAV Produce Chemically and Functionally Distinct Vectors. Mol. Ther. Methods Clin. Dev. 2020, 18, 98–118. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Rosario, A.; Cruz, P.; Siemienski, Z.; Ceballos-Diaz, C.; Crosby, K.; Jansen, K.; Borchelt, D.R.; Kim, J.Y.; Jankowsky, J.L.; et al. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS ONE 2013, 8, e67680. [Google Scholar] [CrossRef]
- Mattar, C.N.; Waddington, S.N.; Biswas, A.; Johana, N.; Ng, X.W.; Fisk, A.S.; Fisk, N.M.; Tan, L.G.; Rahim, A.A.; Buckley, S.M.; et al. Systemic delivery of scAAV9 in fetal macaques facilitates neuronal transduction of the central and peripheral nervous systems. Gene. Ther. 2013, 20, 69–83. [Google Scholar] [CrossRef]
- Gray, S.J.; Matagne, V.; Bachaboina, L.; Yadav, S.; Ojeda, S.R.; Samulski, R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: A comparative study of adult mice and nonhuman primates. Mol. Ther. 2011, 19, 1058–1069. [Google Scholar] [CrossRef]
- Dehay, B.; Dalkara, D.; Dovero, S.; Li, Q.; Bezard, E. Systemic scAAV9 variant mediates brain transduction in newborn rhesus macaques. Sci. Rep. 2012, 2, 253. [Google Scholar] [CrossRef]
- Bevan, A.K.; Duque, S.; Foust, K.D.; Morales, P.R.; Braun, L.; Schmelzer, L.; Chan, C.M.; McCrate, M.; Chicoine, L.G.; Coley, B.D.; et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 2011, 19, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Koch, W.J.; Rabinowitz, J.E. Comparative cardiac gene delivery of adeno-associated virus serotypes 1–9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin. Transl. Sci. 2010, 3, 81–89. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.I.; Li, J.; Sun, L.; Zhang, J.; Xiao, X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene. Ther. 2003, 10, 2105–2111. [Google Scholar] [CrossRef]
- Rincon, M.Y.; de Vin, F.; Duque, S.I.; Fripont, S.; Castaldo, S.A.; Bouhuijzen-Wenger, J.; Holt, M.G. Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene. Ther. 2018, 25, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Lee, H.; Rah, W.; Chang, H.J.; Yoon, Y.S. From engineered heart tissue to cardiac organoid. Theranostics 2022, 12, 2758–2772. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, M. Redirection of Tropism of AAV Capsids. #WO 2021/230987 A1, 31 March 2021. [Google Scholar]
- Rodrigues, G.A.; Shalaev, E.; Karami, T.K.; Cunningham, J.; Slater, N.K.H.; Rivers, H.M. Pharmaceutical Development of AAV-Based Gene Therapy Products for the Eye. Pharm. Res. 2018, 36, 29. [Google Scholar] [CrossRef] [PubMed]
- Au, H.K.E.; Isalan, M.; Mielcarek, M. Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 2021, 8, 809118. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.K.; Khan, N.; Parker, C.L.; Samulski, R.J.; Matsushima, G.; Gray, S.J.; McCown, T.J. Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene. Ther. 2016, 23, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Muller, O.J.; Kaul, F.; Weitzman, M.D.; Pasqualini, R.; Arap, W.; Kleinschmidt, J.A.; Trepel, M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003, 21, 1040–1046. [Google Scholar] [CrossRef]
- Chen, Y.H.; Chang, M.; Davidson, B.L. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat. Med. 2009, 15, 1215–1218. [Google Scholar] [CrossRef]
- Wang, D.; Li, S.; Gessler, D.J.; Xie, J.; Zhong, L.; Li, J.; Tran, K.; Van Vliet, K.; Ren, L.; Su, Q.; et al. A Rationally Engineered Capsid Variant of AAV9 for Systemic CNS-Directed and Peripheral Tissue-Detargeted Gene Delivery in Neonates. Mol. Ther. Methods Clin. Dev. 2018, 9, 234–246. [Google Scholar] [CrossRef]
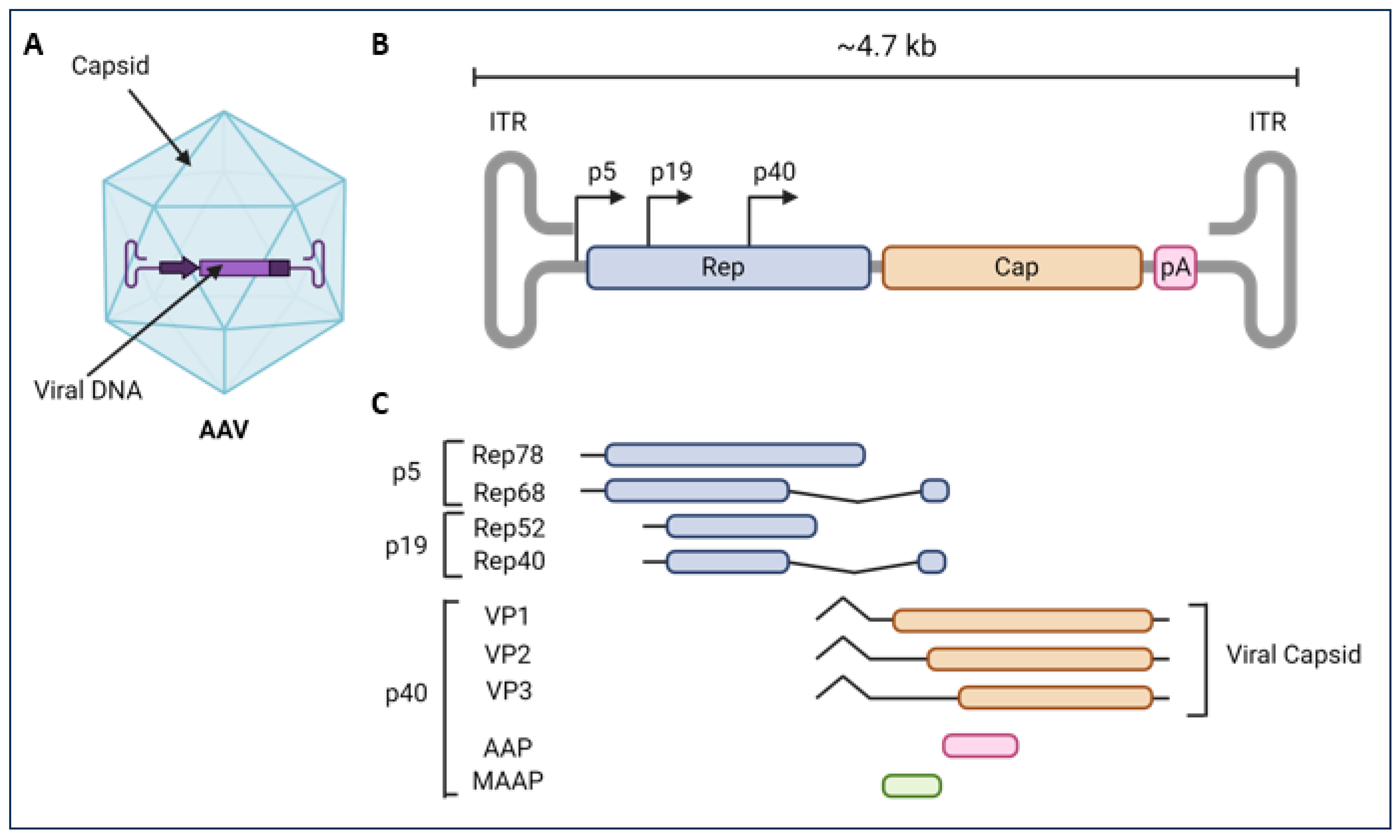

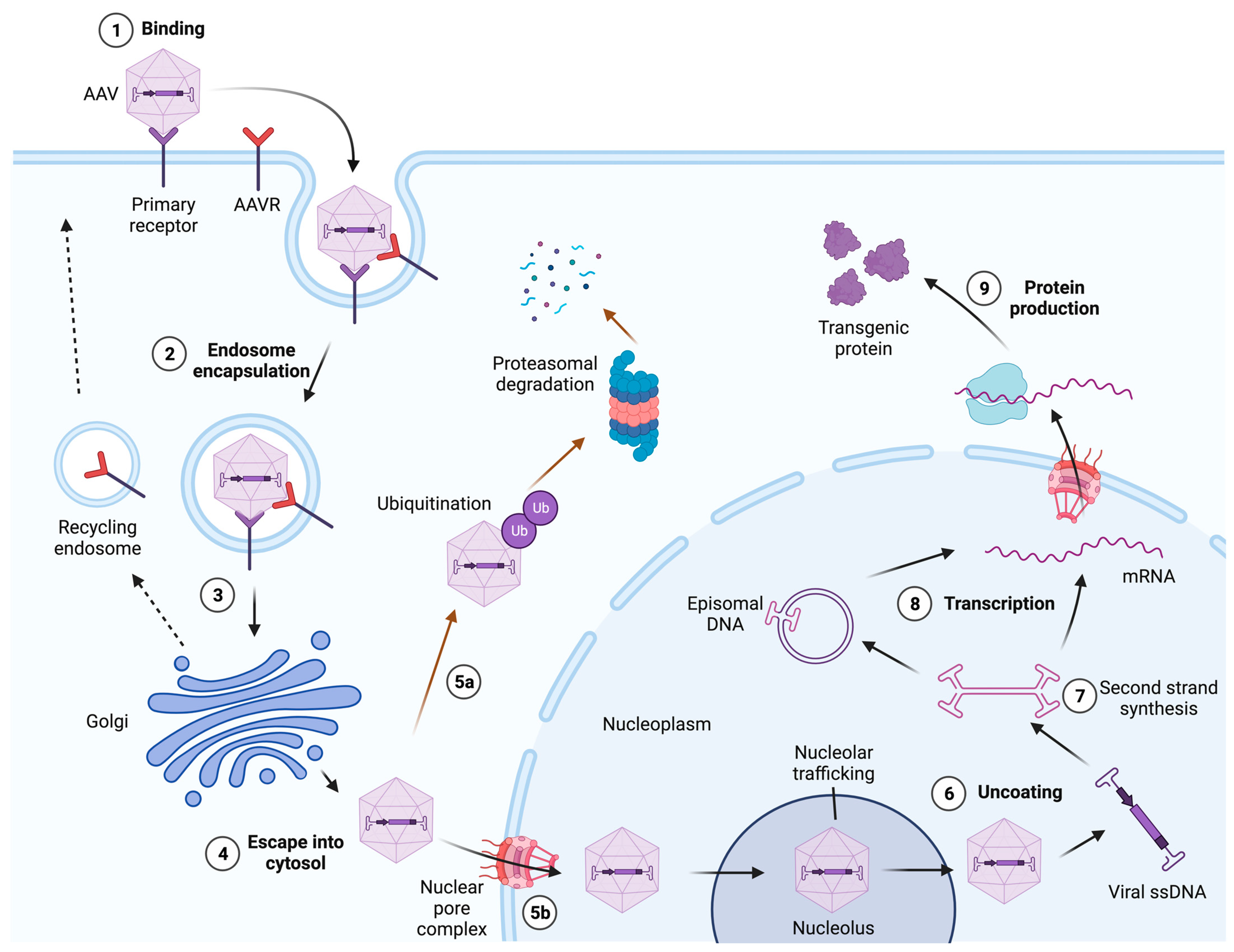
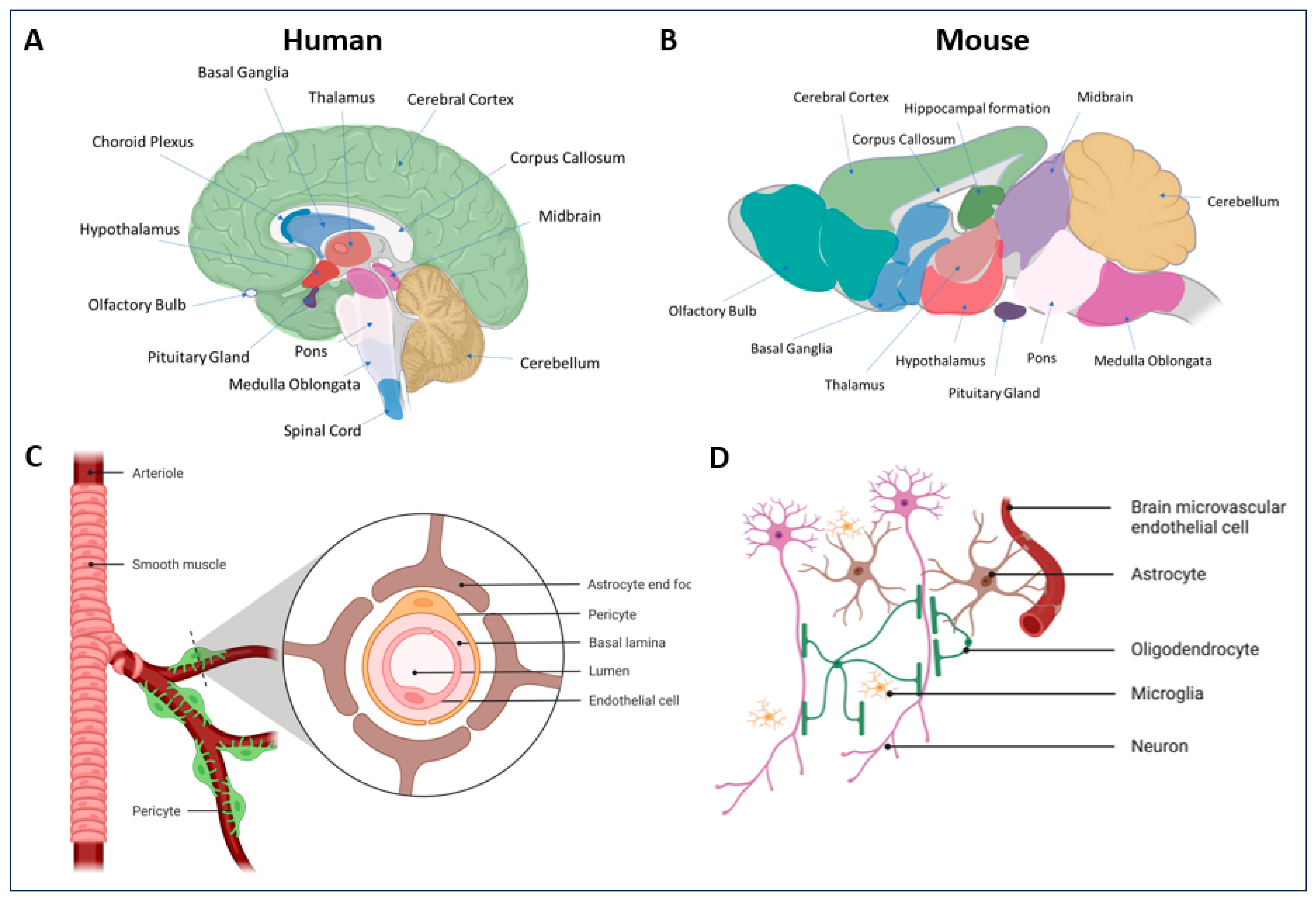
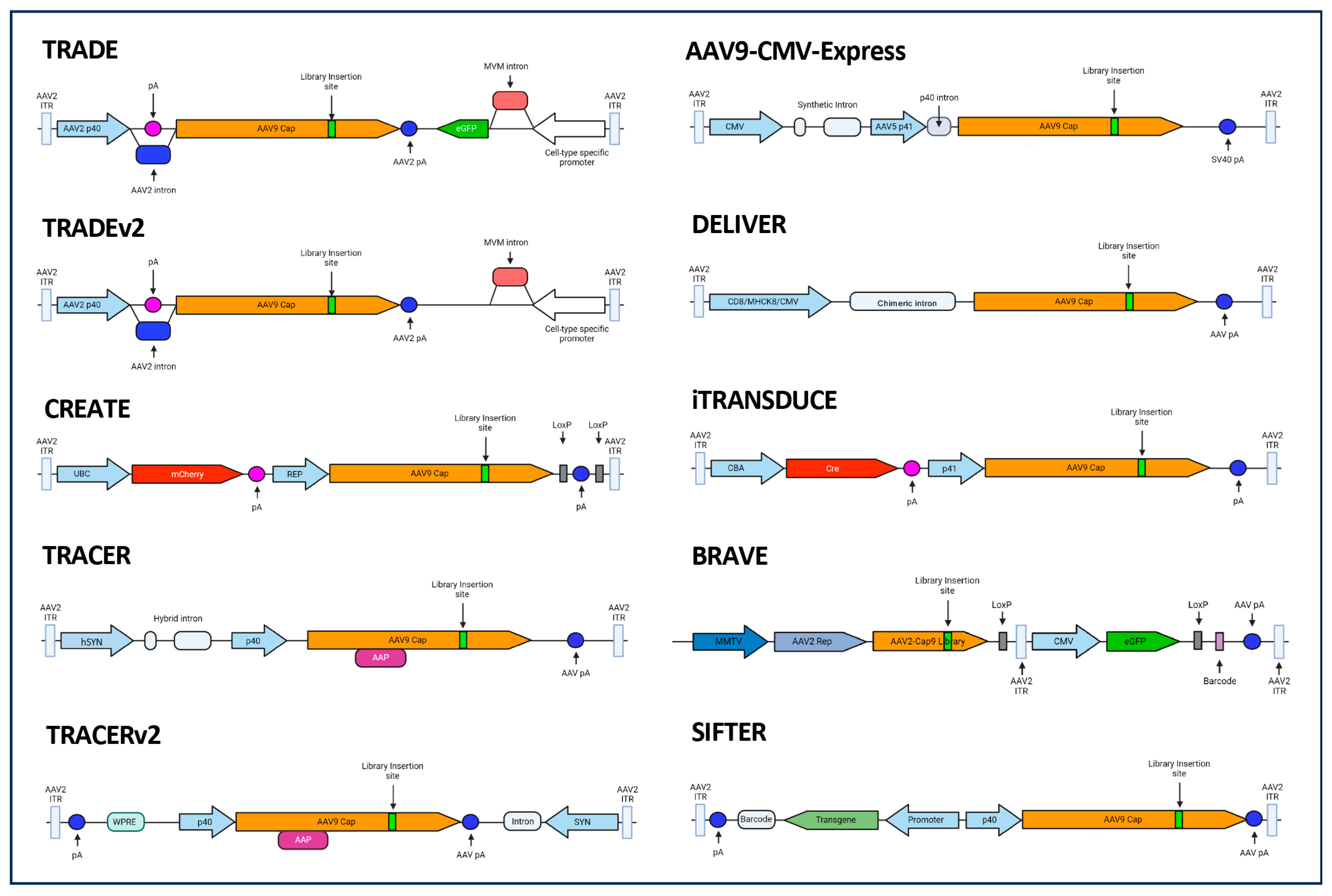

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SEROTYPE | RECEPTOR(S) | PUTATIVE CO-RECEPTOR(S) | REFERENCES |
|---|---|---|---|
| AAV1 | α2-3 N-linked SIA and AAVR | Unknown | [99,107,108] |
| AAV2 | HSPG and AAVR | FGFR1, αVβ5 integrin, α5β1 integrin, HGFR, LR, and CD9 | [58,90,91,92,94,95,96,99,108,109] |
| AAV3 | HSPG and AAVR | FGFR1, HGFR, and LR | [96,99,104,105,106,108,110,111] |
| AAV4 | α2-3 O-linked SIA | Unknown | [112] |
| AAV5 | α2-3 N-linked SIA and AAVR | α and β PDGFR | [99,108,112,113] |
| AAV6 | HSPG, α2-3 and α2-6 N-linked SIA, and AAVR | EGFR | [99,104,105,107,114] |
| AAV7 | Unknown | Unknown | |
| AAV8 | LR and AAVR | Unknown | [96,99,108] |
| AAV9 | Terminal N-linked galactose, and AAVR | LR and putative integrin | [96,99,108,115,116] |
| AAV10 | Unknown | Unknown | |
| AAV11 | Unknown | Unknown | |
| AAV12 | Putative mannose and mannosamine | Unknown | [117] |
| AAV13 | Unknown | HSPG | [104,105] |
| AAVrh.10 | Sulfated N-acetyllactosamine (LacNAc) and AAVR | Unknown | [108,118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Gordo, E.; Chamberlain, K.; Riyad, J.M.; Kohlbrenner, E.; Weber, T. Natural Adeno-Associated Virus Serotypes and Engineered Adeno-Associated Virus Capsid Variants: Tropism Differences and Mechanistic Insights. Viruses 2024, 16, 442. https://doi.org/10.3390/v16030442
Lopez-Gordo E, Chamberlain K, Riyad JM, Kohlbrenner E, Weber T. Natural Adeno-Associated Virus Serotypes and Engineered Adeno-Associated Virus Capsid Variants: Tropism Differences and Mechanistic Insights. Viruses. 2024; 16(3):442. https://doi.org/10.3390/v16030442
Chicago/Turabian StyleLopez-Gordo, Estrella, Kyle Chamberlain, Jalish Mahmud Riyad, Erik Kohlbrenner, and Thomas Weber. 2024. "Natural Adeno-Associated Virus Serotypes and Engineered Adeno-Associated Virus Capsid Variants: Tropism Differences and Mechanistic Insights" Viruses 16, no. 3: 442. https://doi.org/10.3390/v16030442
APA StyleLopez-Gordo, E., Chamberlain, K., Riyad, J. M., Kohlbrenner, E., & Weber, T. (2024). Natural Adeno-Associated Virus Serotypes and Engineered Adeno-Associated Virus Capsid Variants: Tropism Differences and Mechanistic Insights. Viruses, 16(3), 442. https://doi.org/10.3390/v16030442