
Involvement of the Cell Division Protein DamX in the Infection Process of Bacteriophage T4


{kind=link}
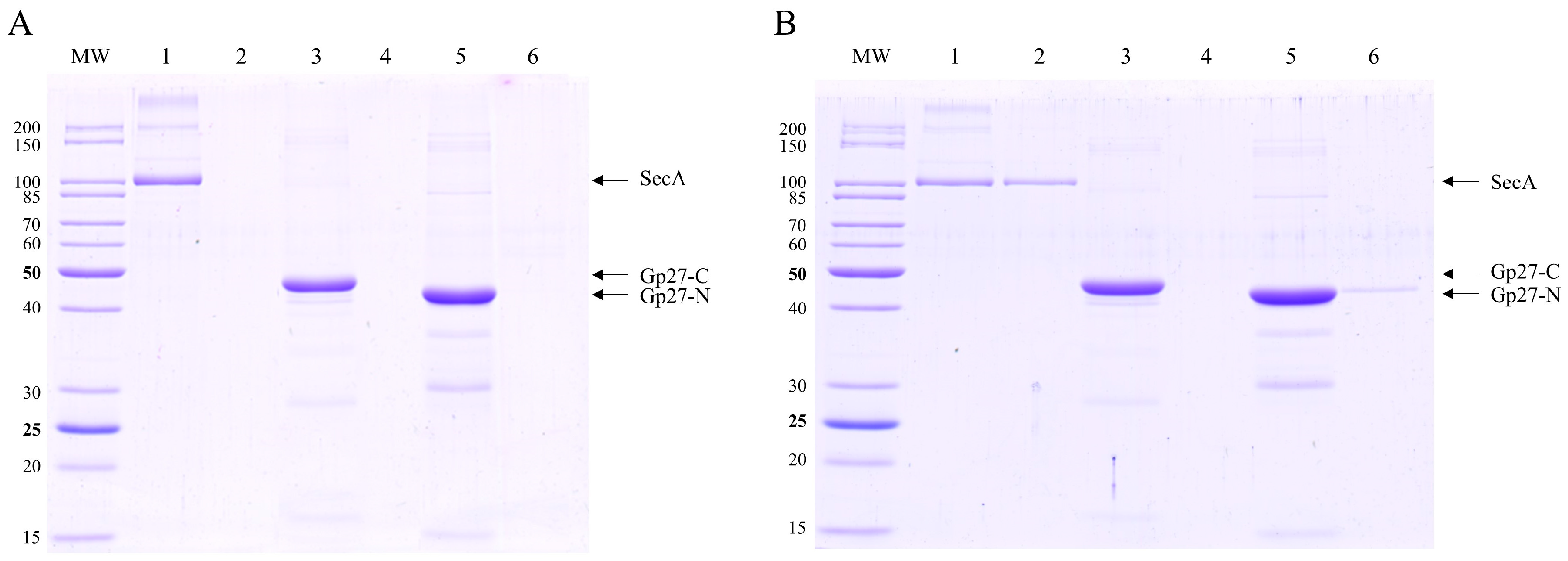{kind=link}
{kind=link}
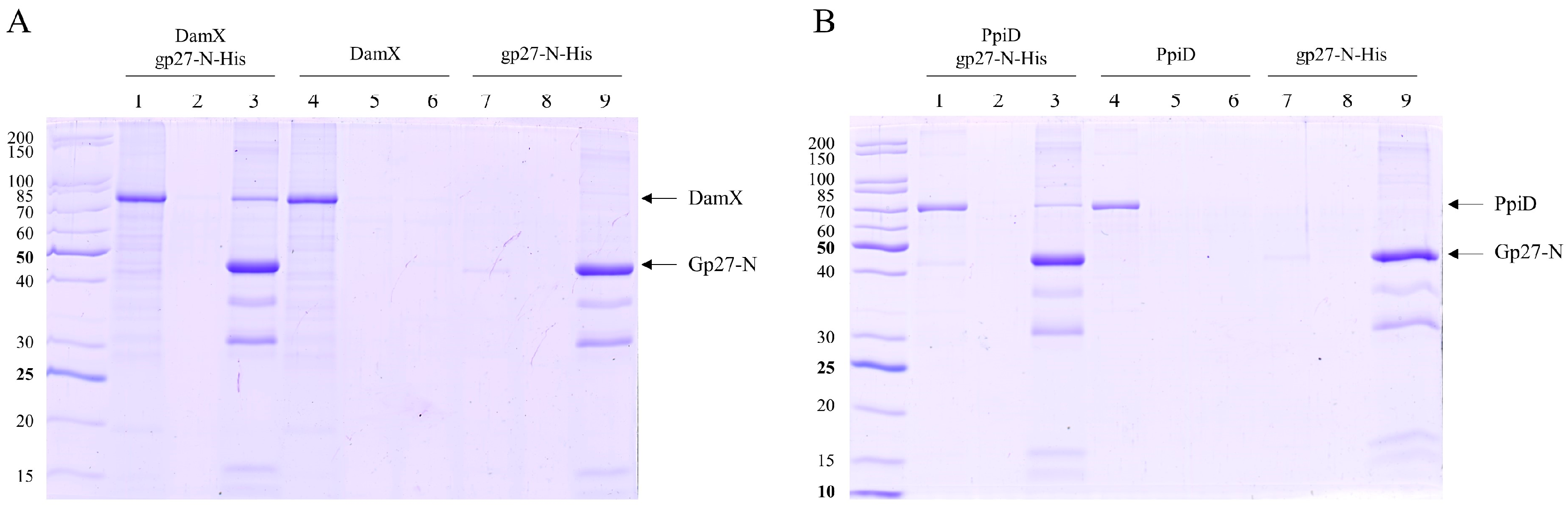{kind=link}
{kind=link}
{kind=link}
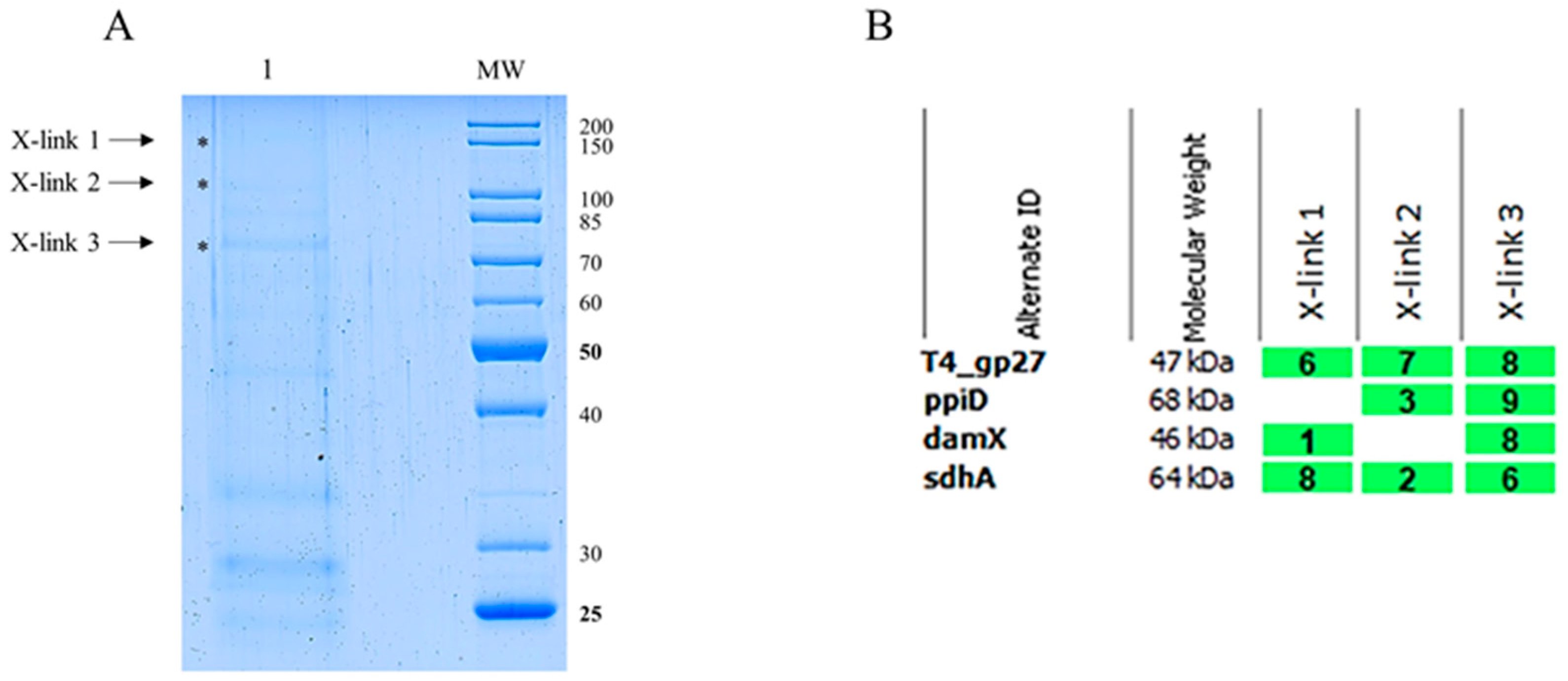{kind=link}
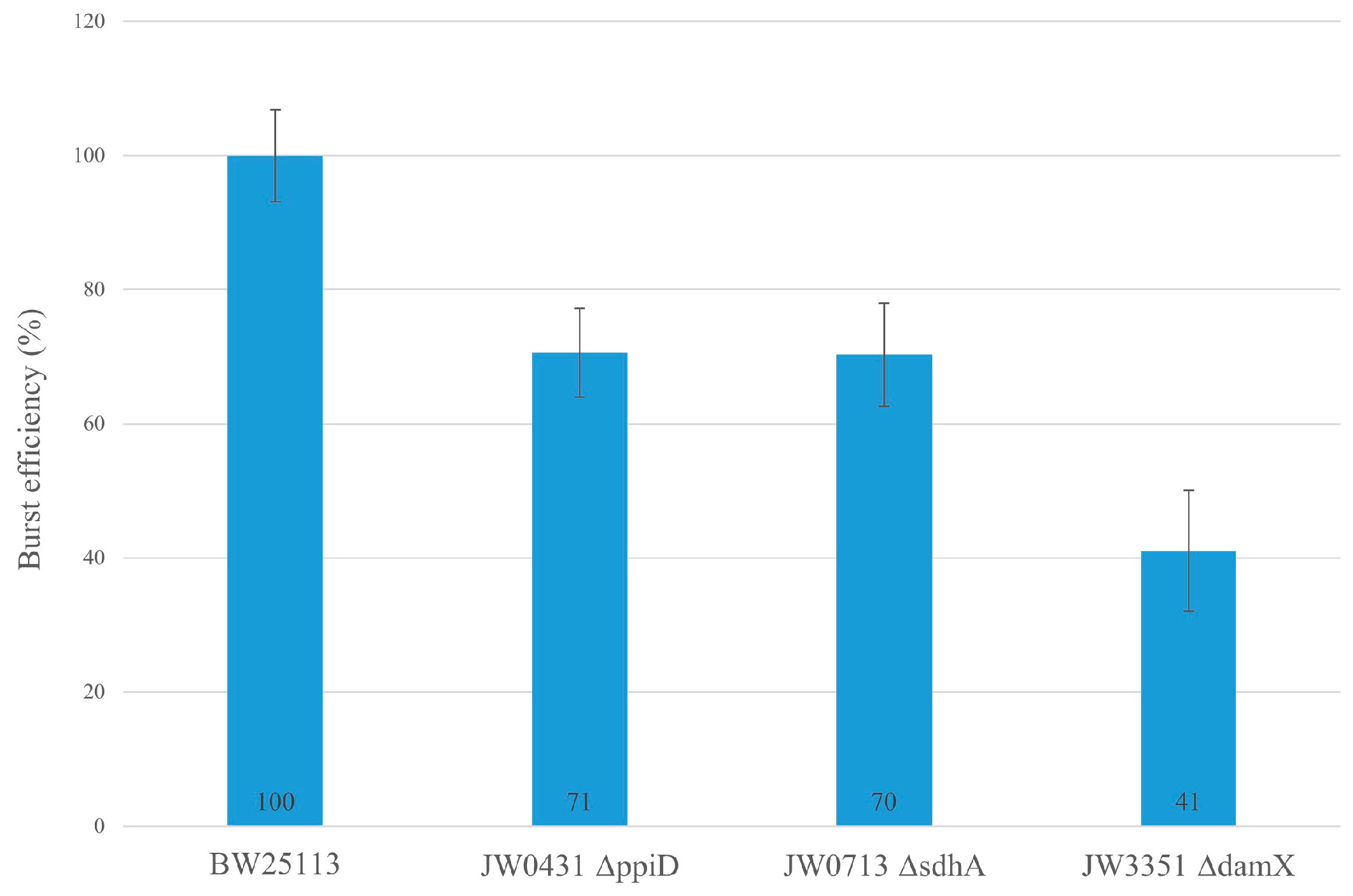{kind=link}
Abstract
Share and Cite
Wenzel, S.; Hess, R.; Kiefer, D.; Kuhn, A. Involvement of the Cell Division Protein DamX in the Infection Process of Bacteriophage T4. Viruses 2024, 16, 487. https://doi.org/10.3390/v16040487
Wenzel S, Hess R, Kiefer D, Kuhn A. Involvement of the Cell Division Protein DamX in the Infection Process of Bacteriophage T4. Viruses. 2024; 16(4):487. https://doi.org/10.3390/v16040487
Chicago/Turabian StyleWenzel, Sabrina, Renate Hess, Dorothee Kiefer, and Andreas Kuhn. 2024. "Involvement of the Cell Division Protein DamX in the Infection Process of Bacteriophage T4" Viruses 16, no. 4: 487. https://doi.org/10.3390/v16040487
APA StyleWenzel, S., Hess, R., Kiefer, D., & Kuhn, A. (2024). Involvement of the Cell Division Protein DamX in the Infection Process of Bacteriophage T4. Viruses, 16(4), 487. https://doi.org/10.3390/v16040487