Deciphering the Role of Epstein–Barr Virus Latent Membrane Protein 1 in Immune Modulation: A Multifaced Signalling Perspective

 , , and
, , and 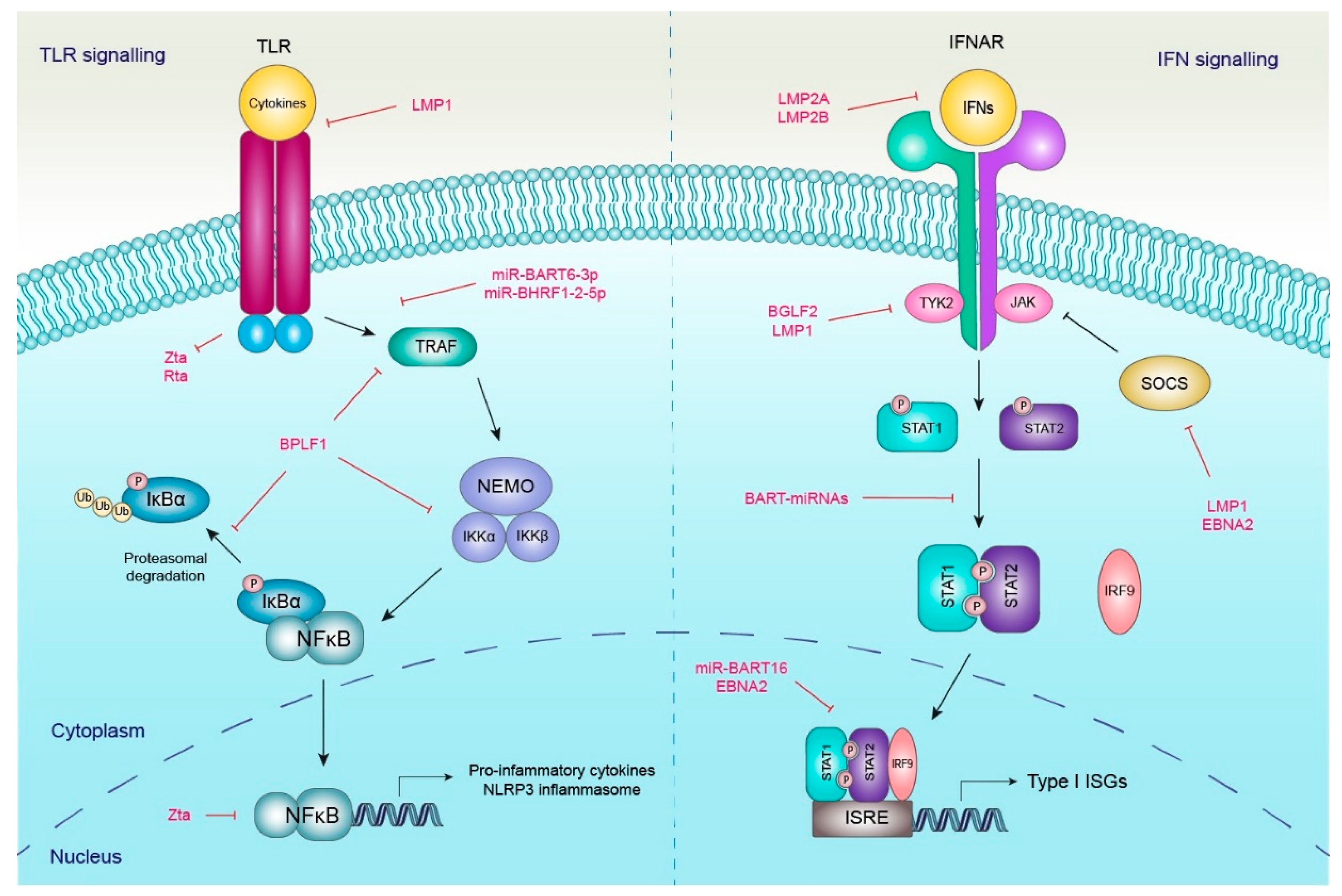{kind=link}
{kind=link}
Abstract
:1. Introduction
EBV Cell Entry
2. Innate Immune Response to Epstein–Barr Virus
2.1. Inflammasome Impact on EBV Infection
2.2. Modulation of Type I IFN Response
3. Specific Immune Response to EBV
CD8+ T Cell Response in Chronic EBV Infection
4. EBV-Mediated Induction of Immunomodulatory Molecules by Activation of Signal Transduction Pathways
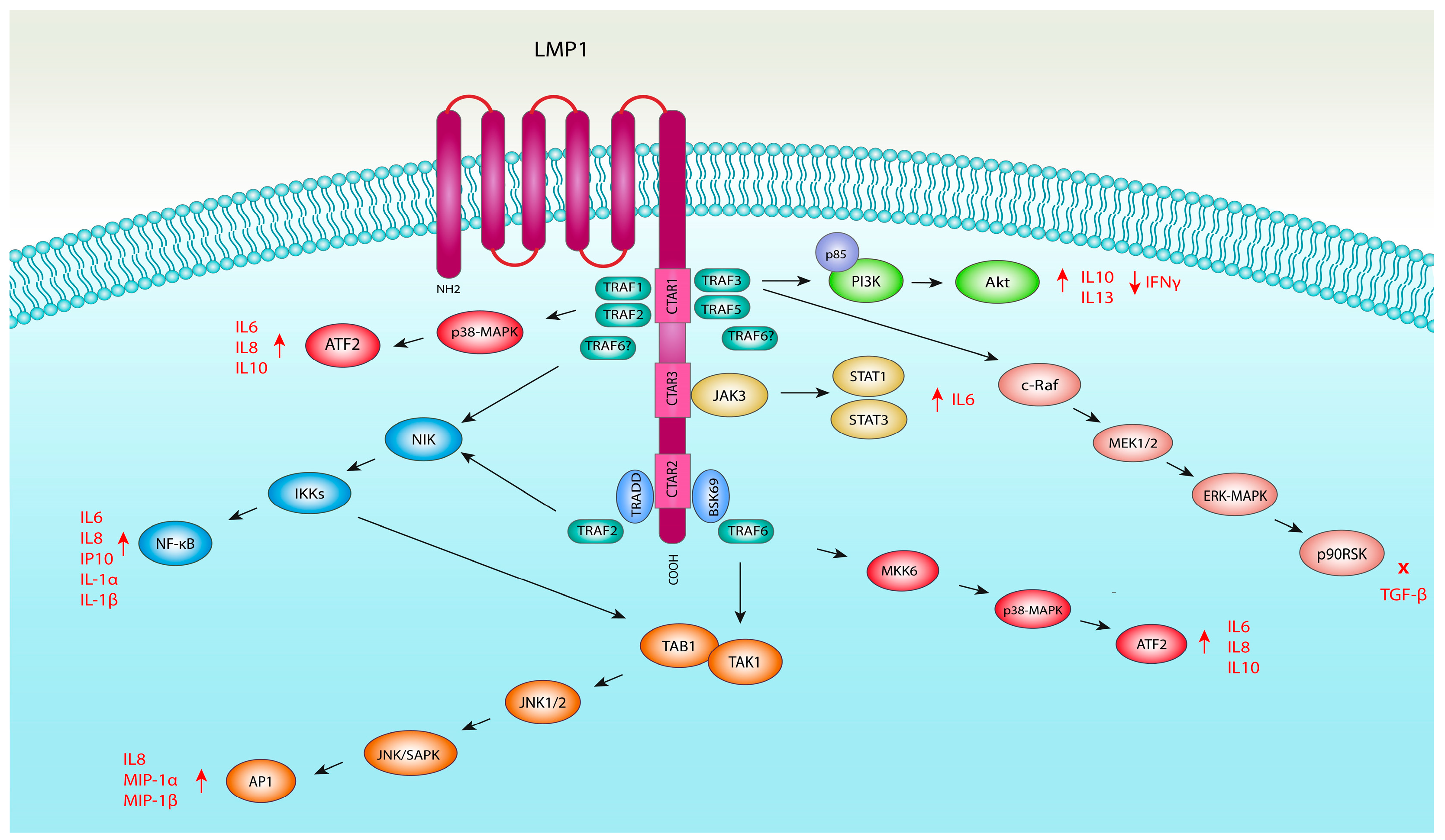
4.1. LMP1 and NF-κB Pathway Activation
4.2. LMP1 and MAPK Pathway Activation
4.2.1. LMP1 and JNK Pathway Activation
4.2.2. LMP1 and ERK1/2 Pathway Activation
4.2.3. LMP1 and p38 Mitogen-Activated Kinase Pathway
4.3. LMP1 and JAK/STAT Signalling Pathway Activation
4.4. LMP1 and PI3K/Akt Pathway Activation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses ICTV (2022): Current ICTV Taxonomy Release: Taxon Details: Epstein-Barr Virus. Available online: https://ictv.global/taxonomy/taxondetails?taxnode_id=19780268 (accessed on 14 January 2024).
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein–Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Rozman, M.; Korać, P.; Jambrosic, K.; Židovec Lepej, S. Progress in Prophylactic and Therapeutic EBV Vaccine Development Based on Molecular Characteristics of EBV Target Antigens. Pathogens 2022, 11, 864. [Google Scholar] [CrossRef]
- Pope, J.H. Establishment of cell lines from peripheral leucocytes in infectious mononucleosis. Nature 1967, 216, 810–811. [Google Scholar] [CrossRef] [PubMed]
- Henle, W.; Diehl, V.; Kohn, G.; Hausen, H.Z.; Henle, G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 1967, 157, 1064–1065. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, W.L.; Zhu, Q.; Zhang, S.; Yao, Y.Y.; Xiang, T.; Feng, Q.S.; Zhang, Z.; Peng, R.J.; Jia, W.H.; et al. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostics 2019, 9, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X.; et al. Targeting the Signaling in Epstein–Barr Virus-Associated Diseases: Mechanism, Regulation, and Clinical Study. Signal Transduct. Target. Ther. 2021, 6, 15. [Google Scholar] [CrossRef]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV Membrane Protein Expressed in Immortalized Lymphocytes Transforms Established Rodent Cells. Cell 1985, 43, 831–840. [Google Scholar] [CrossRef]
- Kaye, K.M.; Izumi, K.M.; Kieff, E. Epstein-Barr Virus Latent Membrane Protein 1 Is Essential for B-Lymphocyte Growth Transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 9150–9154. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent Membrane Protein 1 of Epstein-Barr Virus Mimics a Constitutively Active Receptor Molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar] [CrossRef]
- Chen, J.; Longnecker, R. Epithelial Cell Infection by Epstein–Barr Virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.; Hutt-Fletcher, L.M. Epstein-Barr Virus Enters B Cells and Epithelial Cells by Different Routes. J. Virol. 1992, 66, 3409–3414. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Joyce, M.G.; Nguyen, H.; Banh, D.V.; Aguilar, F.; Tariq, Z.; Yap, M.L.; Tsujimura, Y.; Gillespie, R.A.; Tsybovsky, Y.; et al. Immunization with Components of the Viral Fusion Apparatus Elicits Antibodies That Neutralize Epstein-Barr Virus in B Cells and Epithelial Cells. Immunity 2019, 50, 1305–1316.e6. [Google Scholar] [CrossRef]
- Hong, J.; Wei, D.; Zhong, L.; Wu, Q.; Chen, K.; Zhang, W.; Yang, Y.; Chen, J.; Xia, N.; Zhang, X.; et al. Glycoprotein B Antibodies Completely Neutralize EBV Infection of B Cells. Front. Immunol. 2022, 13, 920467. [Google Scholar] [CrossRef] [PubMed]
- Snijder, J.; Ortego, M.S.; Weidle, C.; Stuart, A.B.; Gray, M.D.; McElrath, M.J.; Pancera, M.; Veesler, D.; McGuire, A.T. An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 2018, 48, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wei, D.; Wu, Q.; Zhong, L.; Chen, K.; Huang, Y.; Zhang, W.; Chen, J.; Xia, N.; Zhang, X.; et al. Antibody Generation and Immunogenicity Analysis of EBV Gp42 N-Terminal Region. Viruses 2021, 13, 2380. [Google Scholar] [CrossRef] [PubMed]
- Bu, G.-L.; Xie, C.; Kang, Y.-F.; Zeng, M.-S.; Sun, C. How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection. Viruses 2022, 14, 2372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin Receptor A2 Is an Epithelial Cell Receptor for Epstein–Barr Virus Entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin Receptor A2 Is a Functional Entry Receptor for Epstein–Barr Virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Wallaschek, N.; Reuter, S.; Silkenat, S.; Wolf, K.; Niklas, C.; Kayisoglu, Ö.; Aguilar, C.; Wiegering, A.; Germer, C.-T.; Kircher, S.; et al. Ephrin Receptor A2, the Epithelial Receptor for Epstein-Barr Virus Entry, Is Not Available for Efficient Infection in Human Gastric Organoids. PLoS Pathog. 2021, 17, e1009210. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.A.; Coleman, C.B.; Gewurz, B.E.; Rochford, R. CD21 (Complement Receptor 2) Is the Receptor for Epstein-Barr Virus Entry into T Cells. J. Virol. 2020, 94, e00428-20. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Chathuranga, K.; Lee, J.-S. Intracellular Sensing of Viral Genomes and Viral Evasion. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Jangra, S.; Yuen, K.-S.; Botelho, M.G.; Jin, D.-Y. Epstein–Barr Virus and Innate Immunity: Friends or Foes? Microorganisms 2019, 7, 183. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Looi, C.K.; Hii, L.-W.; Chung, F.F.-L.; Mai, C.-W.; Lim, W.-M.; Leong, C.-O. Roles of Inflammasomes in Epstein–Barr Virus-Associated Nasopharyngeal Cancer. Cancers 2021, 13, 1786. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.M.; Goldbach-Mansky, R.; Bhaduri-McIntosh, S. A Promiscuous Inflammasome Sparks Replication of a Common Tumor Virus. Proc. Natl. Acad. Sci. USA 2020, 117, 1722–1730. [Google Scholar] [CrossRef]
- Li, Z.; Duan, Y.; Cheng, S.; Chen, Y.; Hu, Y.; Zhang, L.; He, J.; Liao, Q.; Yang, L.; Sun, L.-Q. EBV-Encoded RNA via TLR3 Induces Inflammation in Nasopharyngeal Carcinoma. Oncotarget 2015, 6, 24291–24303. [Google Scholar] [CrossRef]
- Lussignol, M.; Esclatine, A. Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”. Viruses 2017, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The Latent Membrane Protein 1 Oncogene Modifies B-Cell Physiology by Regulating Autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive Autophagy Contributes to Resistance to TP53-Mediated Apoptosis in Epstein-Barr Virus-Positive Latency III B-Cell Lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef]
- Wang, P.; Deng, Y.; Guo, Y.; Xu, Z.; Li, Y.; Ou, X.; Xie, L.; Lu, M.; Zhong, J.; Li, B.; et al. Epstein-Barr Virus Early Protein BFRF1 Suppresses IFN-β Activity by Inhibiting the Activation of IRF3. Front. Immunol. 2020, 11, 513383. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, P.; Barzago, C.; Baggi, F.; Antozzi, C.; Maggi, L.; Mantegazza, R.; Bernasconi, P. Toll-like Receptors 7 and 9 in Myasthenia Gravis Thymus: Amplifiers of Autoimmunity? Ann. N. Y. Acad. Sci. 2018, 1413, 11–24. [Google Scholar] [CrossRef]
- Bouvet, M.; Voigt, S.; Tagawa, T.; Albanese, M.; Chen, Y.A.; Chen, Y.; Fachko, D.N.; Pich, D.; Gobel, C.; Skalsky, R.L.; et al. Multiple Viral microRNAs Regulate Interferon Release and Signaling Early during Infection with Epstein-Barr Virus. mBio 2021, 12, 03440. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus MiR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef]
- Zhang, L.; Pagano, J.S. Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. J. Virol. 2000, 74, 1061–1068. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.-L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-Dependent Viral Recognition by Plasmacytoid Dendritic Cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Severa, M.; Giacomini, E.; Gafa, V.; Anastasiadou, E.; Rizzo, F.; Corazzari, M.; Romagnoli, A.; Trivedi, P.; Fimia, G.M.; Coccia, E.M. EBV stimulates TLR- and autophagy-dependent pathways and impairs maturation in plasmacytoid dendritic cells: Implications for viral immune escape. Eur. J. Immunol. 2013, 43, 147–158. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during Viral Infection—A Double-Edged Sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Santarelli, R.; Granato, M.; Faggioni, A.; Cirone, M. Interference with the Autophagic Process as a Viral Strategy to Escape from the Immune Control: Lesson from Gamma Herpesviruses. J. Immunol. Res. 2015, 2015, 546063. [Google Scholar] [CrossRef]
- Fathallah, I.; Parroche, P.; Gruffat, H.; Zannetti, C.; Johansson, H.; Yue, J.; Manet, E.; Tommasino, M.; Sylla, B.S.; Hasan, U.A. EBV Latent Membrane Protein 1 Is a Negative Regulator of TLR9. J. Immunol. 2010, 185, 6439–6447. [Google Scholar] [CrossRef]
- Xu, C.; Sun, L.; Liu, W.; Duan, Z. Latent Membrane Protein 1 of Epstein–Barr Virus Promotes RIG-I Degradation Mediated by Proteasome Pathway. Front. Immunol. 2018, 9, 1446. [Google Scholar] [CrossRef]
- Ni, Y.; Low, J.T.; Silke, J.; O’Reilly, L.A. Digesting the Role of JAK-STAT and Cytokine Signaling in Oral and Gastric Cancers. Front. Immunol. 2022, 13, 835997. [Google Scholar] [CrossRef]
- Li, Q.; Tan, F.; Wang, Y.; Liu, X.; Kong, X.; Meng, J.; Yang, L.; Cen, S. The Gamble between Oncolytic Virus Therapy and IFN. Front. Immunol. 2022, 13, 971674. [Google Scholar] [CrossRef]
- Li, S.; Gong, M.; Zhao, F.; Shao, J.; Xie, Y.; Zhang, Y.; Chang, H. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell. Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.J.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-Encoded Latent Membrane Proteins, LMP2A and LMP2B, Limit the Actions of Interferon by Targeting Interferon Receptors for Degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef]
- Howe, M.K.; Dowdell, K.; Kuehn, H.S.; Li, Q.; Hart, G.T.; Garabedian, D.; Liepshutz, K.; Hsu, A.P.; Su, H.; Niemela, J.E.; et al. Patients With Natural Killer (NK) Cell Chronic Active Epstein-Barr Virus Have Immature NK Cells and Hyperactivation of PI3K/Akt/MTOR and STAT1 Pathways. J. Infect. Dis. 2020, 222, 1170–1179. [Google Scholar] [CrossRef]
- Liu, X.; Sadaoka, T.; Krogmann, T.; Cohen, J.I. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Suppresses Type I Interferon Signaling To Promote EBV Reactivation. J. Virol. 2020, 94, e00258-20. [Google Scholar] [CrossRef]
- Jangra, S.; Bharti, A.; Lui, W.-Y.; Chaudhary, V.; Botelho, M.G.; Yuen, K.-S.; Jin, D.-Y. Suppression of JAK-STAT Signaling by Epstein-Barr Virus Tegument Protein BGLF2 through Recruitment of SHP1 Phosphatase and Promotion of STAT2 Degradation. J. Virol. 2021, 95, e01027-21. [Google Scholar] [CrossRef]
- Wood, V.H.; O’Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFβ signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Yu, Y.; Ouyang, Y.; Chen, Z.; He, X.; He, Z.-M. Carboxyl Terminal Activating Region 3 of Latent Membrane Protein 1 Encoded by the Epstein-Barr Virus Regulates Cell Proliferation and Protein Expression in NP69 Cells. Mol. Med. Rep. 2019, 21, 720–730. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Pai, L.-M.; Lee, C.-K. Fine-Tuning of Type I Interferon Response by STAT3. Front. Immunol. 2019, 10, 1448. [Google Scholar] [CrossRef]
- Muromoto, R.; Ikeda, O.; Okabe, K.; Togi, S.; Kamitani, S.; Fujimuro, M.; Harada, S.; Oritani, K.; Matsuda, T. Epstein–Barr Virus-Derived EBNA2 Regulates STAT3 Activation. Biochem. Biophys. Res. Commun. 2009, 378, 439–443. [Google Scholar] [CrossRef]
- Kwon, H.J.; Yang, J.M.; Lee, J.-O.; Lee, J.S.; Paik, J.H. Clinicopathologic Implication of PD-L1 and Phosphorylated STAT3 Expression in Diffuse Large B Cell Lymphoma. J. Transl. Med. 2018, 16, 320. [Google Scholar] [CrossRef]
- Sobah, M.L.; Liongue, C.; Ward, A.C. SOCS Proteins in Immunity, Inflammatory Diseases, and Immune-Related Cancer. Front. Med. 2021, 8, 727987. [Google Scholar] [CrossRef]
- Durham, G.A.; Williams, J.J.L.; Nasim, M.T.; Palmer, T.M. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol. Sci. 2019, 40, 298–308. [Google Scholar] [CrossRef]
- Wood, C.D.; Carvell, T.; Gunnell, A.; Ojeniyi, O.O.; Osborne, C.; West, M.J. Enhancer Control of MicroRNA MiR-155 Expression in Epstein-Barr Virus-Infected B Cells. J. Virol. 2018, 92, e00716-18. [Google Scholar] [CrossRef]
- Rex, V.; Zargari, R.; Stempel, M.; Halle, S.; Brinkmann, M.M. The Innate and T-Cell Mediated Immune Response during Acute and Chronic Gammaherpesvirus Infection. Front. Cell. Infect. Microbiol. 2023, 13, 1146381. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Fischer, A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Callan, M.F.C.; Tan, L.; Annels, N.; Ogg, G.S.; Wilson, J.D.K.; O’Callaghan, C.A.; Steven, N.; McMichael, A.J.; Rickinson, A.B. Direct Visualization of Antigen-Specific CD8+ T Cells during the Primary Immune Response to Epstein-Barr Virus In Vivo. J. Exp. Med. 1998, 187, 1395–1402. [Google Scholar] [CrossRef]
- Morvan, M.G.; Teque, F.C.; Locher, C.P.; Levy, J.A. The CD8+ T Cell Noncytotoxic Antiviral Responses. Microbiol. Mol. Biol. Rev. 2021, 85, e00155-20. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Gent, M.; Gram, A.M.; Hooykaas, M.J.; Piersma, S.J.; Wiertz, E.J. Immune Evasion by Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015, 391, 355–381. [Google Scholar] [CrossRef]
- Casco, A.; Johannsen, E. EBV Reactivation from Latency Is a Degrading Experience for the Host. Viruses 2023, 15, 726. [Google Scholar] [CrossRef] [PubMed]
- Almohammed, R.; Osborn, K.; Ramasubramanyan, S.; Perez-Fernandez, I.B.N.; Godfrey, A.; Mancini, E.J.; Sinclair, A.J. Mechanism of Activation of the BNLF2a Immune Evasion Gene of Epstein-Barr Virus by Zta. J. Gen. Virol. 2018, 99, 805–817. [Google Scholar] [CrossRef]
- Fares, S.; Spiess, K.; Olesen, E.T.B.; Zuo, J.; Jackson, S.; Kledal, T.N.; Wills, M.R.; Rosenkilde, M.M. Distinct Roles of Extracellular Domains in the Epstein-Barr Virus-Encoded BILF1 Receptor for Signaling and Major Histocompatibility Complex Class I Downregulation. mBio 2019, 10, e01707-18. [Google Scholar] [CrossRef]
- Lin, J.-H.; Lin, J.-Y.; Chou, Y.-C.; Chen, M.-R.; Yeh, T.-H.; Lin, C.-W.; Lin, S.-J.; Tsai, C.-H. Epstein-Barr Virus LMP2A Suppresses MHC Class II Expression by Regulating the B-Cell Transcription Factors E47 and PU.1. Blood 2015, 125, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Wang, Z.; Ke, Q.; Hong, M.; Qian, Y.; Zhao, X.; Liu, Y.; Kim, H.J.; Ritz, J.; Cantor, H.; et al. Signaling by the Epstein-Barr virus LMP1 protein induces potent cytotoxic CD4+ and CD8+ T cell responses. Proc. Natl. Acad. Sci. USA 2018, 115, E686–E695. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Wakisaka, N.; Crough, T.; Peet, J.; Yoshizaki, T.; Beagley, L.; Khanna, R. Discerning regulation of cis- and trans-presentation of CD8+ T-cell epitopes by EBV-encoded oncogene LMP-1 through self-aggregation. Blood 2009, 113, 6148–6152. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, F.; Tan, H. The Development of CD8 T-Cell Exhaustion Heterogeneity and the Therapeutic Potentials in Cancer. Front. Immunol. 2023, 14, 1166128. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B. The Two Faces of IL-2: A Key Driver of CD8+ T-Cell Exhaustion. Cell. Mol. Immunol. 2021, 18, 1641–1643. [Google Scholar] [CrossRef]
- Dolina, J.S.; Van Braeckel-Budimir, N.; Thomas, G.D.; Salek-Ardakani, S. CD8+ T Cell Exhaustion in Cancer. Front. Immunol. 2021, 12, 715234. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Melese, E.S.; Franks, E.; Cederberg, R.A.; Harbourne, B.T.; Shi, R.; Wadsworth, B.J.; Collier, J.L.; Halvorsen, E.C.; Johnson, F.; Luu, J.; et al. CCL5 Production in Lung Cancer Cells Leads to an Altered Immune Microenvironment and Promotes Tumor Development. OncoImmunology 2022, 11, 2010905. [Google Scholar] [CrossRef]
- Martinez, O.M.; Krams, S.M. The Immune Response to Epstein Barr Virus and Implications for Posttransplant Lymphoproliferative Disorder. Transplantation 2017, 101, 2009–2016. [Google Scholar] [CrossRef]
- Salnikov, M.; Prusinkiewicz, M.A.; Lin, S.; Ghasemi, F.; Cecchini, M.J.; Mymryk, J.S. Tumor-Infiltrating T Cells in EBV-Associated Gastric Carcinomas Exhibit High Levels of Multiple Markers of Activation, Effector Gene Expression, and Exhaustion. Viruses 2023, 15, 176. [Google Scholar] [CrossRef]
- Albanese, M.; Tagawa, T.; Hammerschmidt, W. Strategies of Epstein-Barr virus to evade innate antiviral immunity of its human host. Front. Microbiol. 2022, 13, 955603. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.L.; Martinez, O.M. Latent Membrane Protein 1 of EBV Activates Phosphatidylinositol 3-Kinase to Induce Production of IL-10. J. Immunol. 2007, 179, 8225–8234. [Google Scholar] [CrossRef]
- Incrocci, R.; Barse, L.; Stone, A.; Vagvala, S.; Montesano, M.; Subramaniam, V.; Swanson-Mungerson, M. Epstein-Barr Virus Latent Membrane Protein 2A (LMP2A) Enhances IL-10 Production through the Activation of Bruton’s Tyrosine Kinase and STAT3. Virology 2017, 500, 96–102. [Google Scholar] [CrossRef]
- Schönrich, G.; Abdelaziz, M.O.; Raftery, M.J. Epstein-Barr virus, interleukin-10 and multiple sclerosis: A ménage à trois. Front. Immunol. 2022, 13, 1028972. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampé, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B Lymphocytes by Cellular and Epstein-Barr Virus–Encoded Interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 Directly Inhibits CD8+ T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 2018, 48, 299–312.e5. [Google Scholar] [CrossRef]
- Murer, A.; Rühl, J.; Zbinden, A.; Capaul, R.; Hammerschmidt, W.; Chijioke, O.; Münz, C. MicroRNAs of Epstein-Barr Virus Attenuate T-Cell-Mediated Immune Control In Vivo. mBio 2019, 10, e01941-18. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Juszczynski, P.; Rodig, S.J.; Green, M.R.; O’Donnell, E.; Currie, T.; Armant, M.; Takeyama, K.; Monti, S.; Rabinovich, G.A.; et al. Viral Induction and Targeted Inhibition of Galectin-1 in EBV+ Posttransplant Lymphoproliferative Disorders. Blood 2011, 117, 4315–4322. [Google Scholar] [CrossRef]
- Abou Harb, M.; Meckes, D.G.; Sun, L. Epstein-Barr Virus LMP1 Enhances Levels of Large Extracellular Vesicle-Associated PD-L1. J. Virol. 2023, 97, e00219-23. [Google Scholar] [CrossRef]
- Rancan, C.; Schirrmann, L.; Hüls, C.; Zeidler, R.; Moosmann, A. Latent Membrane Protein LMP2A Impairs Recognition of EBV-Infected Cells by CD8+ T Cells. PLoS Pathog. 2015, 11, e1004906. [Google Scholar] [CrossRef] [PubMed]
- Wasil, L.R.; Tomaszewski, M.J.; Hoji, A.; Rowe, D.T. The Effect of Epstein-Barr Virus Latent Membrane Protein 2 Expression on the Kinetics of Early B Cell Infection. PLoS ONE 2013, 8, e54010. [Google Scholar] [CrossRef]
- Portis, T.; Ikeda, M.; Longnecker, R. Epstein–Barr Virus LMP2A: Regulating Cellular Ubiquitination Processes for Maintenance of Viral Latency? Trends Immunol. 2004, 25, 422–426. [Google Scholar] [CrossRef]
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 2012, 22, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Zhu, J.; Zhao, Q.; Ma, W.; Xiao, Y.; Xu, G.; Zhang, Z. LMP1 Up-regulates Calreticulin to Induce Epithelial-mesenchymal Transition via TGF-β/Smad3/NRP1 Pathway in Nasopharyngeal Carcinoma Cells. J. Cancer 2020, 11, 1257–1269. [Google Scholar] [CrossRef]
- Aviel, S.; Winberg, G.; Massucci, M.; Ciechanover, A. Degradation of the Epstein-Barr Virus Latent Membrane Protein 1 (LMP1) by the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 2000, 275, 23491–23499. [Google Scholar] [CrossRef]
- Coffin III, W.F.; Erickson, K.D.; Hoedt-Miller, M.; Martin, J.M. The cytoplasmic amino-terminus of the Latent Membrane Protein-1 of Epstein-Barr Virus: Relationship between transmembrane orientation and effector functions of the carboxy-terminus and transmembrane domain. Oncogene 2001, 20, 5313–5330. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.P.; Chen, C.C.; Wu, C.C.; Huang, Y.C.; Liu, S.C.; Liang, Y.; Chang, K.P.; Chang, Y.S. Epstein-Barr Virus-Encoded LMP1 Interacts with FGD4 to Activate Cdc42 and Thereby Promote Migration of Nasopharyngeal Carcinoma Cells. PLoS Pathog. 2012, 8, e1002690. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein–Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-κB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, L.; Hong, K.; Pagano, J.S. Intracellular Signaling Molecules Activated by Epstein-Barr Virus for Induction of Interferon Regulatory Factor 7. J. Virol. 2001, 75, 12393–12401. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, W.; Wu, L.; Bai, T.; Zhang, M.; Lo, K.W.; Chui, Y.L.; Cui, Y.; Tao, Q.; Yamamoto, M.; et al. BS69, a Specific Adaptor in the Latent Membrane Protein 1-Mediated c-Jun N-Terminal Kinase Pathway. Mol. Cell. Biol. 2006, 26, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Kieser, A. Pursuing different ‘TRADDes’: TRADD signaling induced by TNF-receptor 1 and the Epstein-Barr virus oncoprotein LMP1. Biol. Chem. 2008, 389, 1261–1271. [Google Scholar] [CrossRef]
- Cuomo, L.; Ramquist, T.; Trivedi, P.; Wang, F.; Klein, G.; Masucci, M.G. Expression of the Epstein-barr virus (EBV)-encoded membrane protein LMP1 impairs the In vitro growth, clonability and tumorigenicity of an EBV-negative burkitt lymphoma line. Int. J. Cancer 1992, 51, 949–955. [Google Scholar] [CrossRef]
- Laherty, C.; Hu, H.; Opipari, A.; Wang, F.; Dixit, V. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J. Biol. Chem. 1992, 267, 24157–24160. [Google Scholar] [CrossRef] [PubMed]
- Kieser, A.; Kilger, E.; Gires, O.; Ueffing, M.; Kolch, W.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J. 1997, 16, 6478–6485. [Google Scholar] [CrossRef]
- Roberts, M.; Cooper, N.R. Activation of a Ras–MAPK-Dependent Pathway by Epstein–Barr Virus Latent Membrane Protein 1 Is Essential for Cellular Transformation. Virology 1998, 240, 93–99. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Gallagher, N.J.; Blake, S.M.; Dawson, C.W.; Young, L.S. Activation of the p38 Mitogen-activated Protein Kinase Pathway by Epstein-Barr Virus-encoded Latent Membrane Protein 1 Coregulates Interleukin-6 and Interleukin-8 Production. J. Biol. Chem. 1999, 274, 16085–16096. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; Tramountanis, G.; Eliopoulos, A.G.; Young, L.S. Epstein-Barr Virus Latent Membrane Protein 1 (LMP1) Activates the Phosphatidylinositol 3-Kinase/Akt Pathway to Promote Cell Survival and Induce Actin Filament Remodeling. J. Biol. Chem. 2003, 278, 3694–3704. [Google Scholar] [CrossRef]
- Lai, H.C.; Hsiao, J.R.; Chen, C.W.; Wu, S.Y.; Lee, C.H.; Su, I.J.; Takada, K.; Chang, Y. Endogenous latent membrane protein 1 in Epstein–Barr virus-infected nasopharyngeal carcinoma cells attracts T lymphocytes through upregulation of multiple chemokines. Virology 2010, 405, 464–473. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Scheidereit, C. IκB kinase complexes: Gateways to NF-κB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a Second, Evolutionary Conserved, NF-κB Signaling Pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The Role of Interleukin 6 during Viral Infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef]
- Ren, Q.; Sato, H.; Murono, S.; Furukawa, M.; Yoshizaki, T. Epstein-Barr Virus (EBV) Latent Membrane Protein 1 Induces Interleukin-8 through the Nuclear Factor-Kappa B Signaling Pathway in EBV-Infected Nasopharyngeal Carcinoma Cell Line. Laryngoscope 2004, 114, 855–859. [Google Scholar] [CrossRef]
- Vockerodt, M.; Pinkert, D.; Smola-Hess, S.; Michels, A.; Ransohoff, R.M.; Tesch, H.; Kube, D. The Epstein-Barr virus oncoprotein latent membrane protein 1 induces expression of the chemokine IP-10: Importance of mRNA half-life regulation. Int. J. Cancer 2005, 114, 598–605. [Google Scholar] [CrossRef]
- Hsu, M.-C.; Wu, S.-Y.; Chang, S.-S.; Su, I.-J.; Tsai, C.-H.; Lai, S.-J.; Shiau, A.-L.; Takada, K.; Chang, Y. Epstein-Barr Virus Lytic Transactivator Zta Enhances Chemotactic Activity through Induction of Interleukin-8 in Nasopharyngeal Carcinoma Cells. J. Virol. 2008, 82, 3679–3688. [Google Scholar] [CrossRef]
- Watanabe, N.; Nodomi, K.; Koike, R.; Kato, A.; Takeichi, O.; Kotani, A.; Kaneko, T.; Sakagami, H.; Takei, M.; Ogata, Y.; et al. EBV LMP1 in Gingival Epithelium Potentially Contributes to Human Chronic Periodontitis Via Inducible IL8 Production. Vivo 2019, 33, 1793–1800. [Google Scholar] [CrossRef]
- Huang, Y.T.; Liu, M.Y.; Tsai, C.H.; Yeh, T.H. Upregulation of interleukin-1 by Epstein-Barr virus latent membrane protein 1 and its possible role in nasopharyngeal carcinoma cell growth. Head Neck 2010, 32, 869–876. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Towfic, F.; Mar, J.C.; Shinners, N.P.; Takasaki, K.; Zhao, B.; Cahir-McFarland, E.D.; Quackenbush, J.; Xavier, R.J.; Kieff, E. Genome-wide siRNA screen for mediators of NF-κB activation. Proc. Natl. Acad. Sci. USA 2012, 109, 2467–2472. [Google Scholar] [CrossRef]
- Ohashi, A.; Uemura, Y.; Yoshimori, M.; Wada, N.; Imadome, K.-I.; Yudo, K.; Koyama, T.; Shimizu, N.; Nishio, M.; Arai, A. The Plasma Level of Interleukin-1β Can Be a Biomarker of Angiopathy in Systemic Chronic Active Epstein–Barr Virus Infection. Front. Microbiol. 2022, 13, 874998. [Google Scholar] [CrossRef] [PubMed]
- Takada, H.; Imadome, K.-I.; Shibayama, H.; Yoshimori, M.; Wang, L.; Saitoh, Y.; Uota, S.; Shoji, Y.; Koyama, T.; Shimizu, N.; et al. EBV Induces Persistent NF-ΚB Activation and Contributes to Survival of EBV-Positive Neoplastic T- or NK-Cells. PLoS ONE 2017, 12, e0174136. [Google Scholar]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.K.; Rashid, F.; Bragg, J.; Ibdah, J.A. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Waites, E.R.; Blake, S.M.S.; Davies, C.; Murray, P.; Young, L.S. TRAF1 Is a Critical Regulator of JNK Signaling by the TRAF-Binding Domain of the Epstein-Barr Virus-Encoded Latent Infection Membrane Protein 1 but Not CD40. J. Virol. 2003, 77, 1316–1328. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yuan, W.; Lin, Z. Functional roles in cell signaling of adaptor protein TRADD from a structural perspective. Comput. Struct. Biotechnol. J. 2020, 18, 2867–2876. [Google Scholar] [CrossRef]
- Izumi, K.M.; McFarland, E.C.; Ting, A.T.; Riley, E.A.; Seed, B.; Kieff, E.D. The Epstein-Barr Virus Oncoprotein Latent Membrane Protein 1 Engages the Tumor Necrosis Factor Receptor-Associated Proteins TRADD and Receptor-Interacting Protein (RIP) but Does Not Induce Apoptosis or Require RIP for NF-κB Activation. Mol. Cell. Biol. 1999, 19, 5759–5767. [Google Scholar] [CrossRef]
- Li, H.P.; Chang, Y.S. Epstein-barr virus latent membrane protein 1: Structure and functions. J. Biomed. Sci. 2003, 10, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Sun, L.; Mendoza, J.W.; Chui, Y.L.; Huang, D.P.; Chen, Z.J.; Suzuki, N.; Suzuki, S.; Yeh, W.C.; Akira, S.; et al. Elucidation of the c-Jun N-Terminal Kinase Pathway Mediated by Epstein-Barr Virus-Encoded Latent Membrane Protein 1. Mol. Cell. Biol. 2004, 24, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Voigt, S.; Sterz, K.R.; Giehler, F.; Mohr, A.-W.; Wilson, J.B.; Moosmann, A.; Kieser, A. A Central Role of IKK2 and TPL2 in JNK Activation and Viral B-Cell Transformation. Nat. Commun. 2020, 11, 685. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, I.; Miller, C.S.; Al-Sabbagh, M. Macrophage Inflammatory Protein-1 Alpha (MIP-1 alpha)/CCL3: As a Biomarker. Biomark. Dis. Methods Discov. Appl. 2015, 223–249. [Google Scholar] [CrossRef]
- Mainou, B.A.; Everly, D.N.; Raab-Traub, N. Unique Signaling Properties of CTAR1 in LMP1-Mediated Transformation. J. Virol. 2007, 81, 9680–9692. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; Laverick, L.; Morris, M.A.; Tramoutanis, G.; Young, L.S. Epstein-Barr Virus-Encoded LMP1 Regulates Epithelial Cell Motility and Invasion via the ERK-MAPK Pathway. J. Virol. 2008, 82, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK–RAS–RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef]
- Guo, Y.; Kamara, S.; Zhang, J.; Wen, H.; Zheng, M.; Liu, Y.; Zhou, L.; Chen, J.; Zhu, S.; Zhang, L. EBV LMP1-C terminal binding affibody molecule downregulates MEK/ERK/p90RSK pathway and inhibits the proliferation of nasopharyngeal carcinoma cells in mouse tumor xenograft models. Front. Cell. Infect. Microbiol. 2023, 12, 1078504. [Google Scholar] [CrossRef]
- Fukuda, M.; Kurosaki, W.; Yanagihara, K.; Kuratsune, H.; Sairenji, T. A Mechanism in Epstein–Barr Virus Oncogenesis: Inhibition of Transforming Growth Factor-β1-mediated Induction of MAPK/p21 by LMP1. Virology 2002, 302, 310–320. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition. J. Signal Transduct. 2012, 2012, 289243. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.A.; Dawson, C.W.; Laverick, L.; Davis, A.M.; Dudman, J.P.R.; Raveenthiraraj, S.; Ahmad, Z.; Yap, L.F.; Young, L.S. The Epstein-Barr virus encoded LMP1 oncoprotein modulates cell adhesion via regulation of activin A/TGFβ and β1 integrin signalling. Sci. Rep. 2016, 6, 19533. [Google Scholar] [CrossRef]
- Morris, M.; Laverick, L.; Wei, W.; Davis, A.; O’Neill, S.; Wood, L.; Wright, J.; Dawson, C.; Young, L. The EBV-Encoded Oncoprotein, LMP1, Induces an Epithelial-to-Mesenchymal Transition (EMT) via Its CTAR1 Domain through Integrin-Mediated ERK-MAPK Signalling. Cancers 2018, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.M.; Rapley, A.; Dawson, C.W.; Young, L.S.; Morris, M.A. The EBV-Encoded Oncoprotein, LMP1, Recruits and Transforms Fibroblasts via an ERK-MAPK-Dependent Mechanism. Pathogens 2021, 10, 982. [Google Scholar] [CrossRef]
- Lo, A.K.F.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1-AMPK pathway by the Epstein-Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Schultheiss, U.; Püschner, S.; Kremmer, E.; Mak, T.W.; Engelmann, H.; Hammerschmidt, W.; Kieser, A. TRAF6 is a critical mediator of signal transduction by the viral oncogene latent membrane protein 1. EMBO J. 2001, 20, 5678–5691. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Jansson, A.; Rüetschi, U.; Rymo, L. The p38 Signaling Pathway Upregulates Expression of the Epstein-Barr Virus LMP1 Oncogene. J. Virol. 2010, 84, 2787–2797. [Google Scholar] [CrossRef]
- Vockerodt, M.; Haier, B.; Buttgereit, P.; Tesch, H.; Kube, D. The Epstein–Barr Virus Latent Membrane Protein 1 Induces Interleukin-10 in Burkitt’s Lymphoma Cells but Not in Hodgkin’s Cells Involving the p38/SAPK2 Pathway. Virology 2001, 280, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Kempuraj, D.; Kandere, K.; Gioacchino, M.D.; Barbacane, R.C.; Castellani, M.L.; Felaco, M.; Boucher, W.; Letourneau, R.; Theoharides, T.C. IL-10, an inflammatory/inhibitory cytokine, but not always. Immunol. Lett. 2003, 86, 123–129. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Beatty, P.R.; Krams, S.M.; Martinez, O.M. Involvement of IL-10 in the autonomous growth of EBV-transformed B cell lines. J. Immunol. 1997, 158, 4045–4051. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Takahara, M.; Nagy, N.; Klein, G.; Klein, E. IL-10 can induce the expression of EBV-encoded latent membrane protein-1 (LMP-1) in the absence of EBNA-2 in B lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell lines. Blood 2006, 107, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Lee, D.C.; Law, A.H.; Fang, J.W.; Chua, D.T.; Lau, A.S. A role for protein kinase PKR in the mediation of Epstein–Barr virus latent membrane protein-1-induced IL-6 and IL-10 expression. Cytokine 2010, 50, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Savitri, E.; Haryana, M.S. Expression of Interleukin-8, Interleukin-10 and Epstein-Barr Viral-Load as Prognostic Indicator in Nasopharyngeal Carcinoma. Glob. J. Health Sci. 2015, 7, 364. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, X.; Zhou, Z.; Li, B.; Peng, J.; Wu, X.; Luo, X.; Yang, L. LMP1-Positive Extracellular Vesicles Promote Radioresistance in Nasopharyngeal Carcinoma Cells through P38 MAPK Signaling. Cancer Med. 2019, 8, 6082–6094. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A Positive Autoregulatory Loop of LMP1 Expression and STAT Activation in Epithelial Cells Latently Infected with Epstein-Barr Virus. J. Virol. 2003, 77, 4139–4148. [Google Scholar] [CrossRef]
- Liu, Y.P.; Tan, Y.N.; Wang, Z.L.; Zeng, L.; Lu, Z.X.; Li, L.L.; Luo, W.; Tang, M.; Cao, Y. Phosphorylation and nuclear translocation of STAT3 regulated by the Epstein-Barr virus latent membrane protein 1 in nasopharyngeal carcinoma. Int. J. Mol. Med. 2008, 21, 153–162. [Google Scholar] [CrossRef]
- Klein, S.C.; Jücker, M.; Abts, H.; Tesch, H. IL6 and IL6 receptor expression in Burkitt’s lymphoma and lymphoblastoid cell lines: Promotion of IL6 receptor expression by EBV. Hematol. Oncol. 1995, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Narazaki, M.; Metwally, H.; Kishimoto, T. Historical overview of the interleukin-6 family cytokine. J. Exp. Med. 2020, 217, e20190347. [Google Scholar] [CrossRef]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorganic Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Lo, A.K.F.; Lo, K.W.; Tsao, S.W.; Wong, H.L.; Hui, J.W.Y.; To, K.F.; Hayward, S.D.; Chui, Y.L.; Lau, Y.L.; Takada, K.; et al. Epstein-Barr Virus Infection Alters Cellular Signal Cascades in Human Nasopharyngeal Epithelial Cells. Neoplasia 2006, 8, 173–180. [Google Scholar]
- Mao, Y.; Wang, J.; Zhang, M.; Fan, W.; Tang, Q.; Xiong, S.; Tang, X.; Xu, J.; Wang, L.; Yang, S.; et al. A neutralized human LMP1-IgG inhibits ENKTL growth by suppressing the JAK3/STAT3 signaling pathway. Oncotarget 2016, 8, 10954–10965. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Chang, C.H.; Huang, Z.N.; Su, Y.C.; Chang, S.J.; Jan, J.S. The JAK Inhibitor Antcin H Exhibits Direct Anticancer Activity While Enhancing Chemotherapy against LMP1-Expressed Lymphoma. Leuk. Lymphoma 2018, 60, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Turner, K.M.; Alfred Yung, W.K.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Liu, M.T.; Chang, Y.T.; Wu, C.C.; Hu, C.Y.; Chen, J.Y. Epstein-Barr Virus Latent Membrane Protein 1 Represses DNA Repair through the PI3K/Akt/FOXO3a Pathway in Human Epithelial Cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Aravinth, S.P.; Rajendran, S.; Li, Y.; Wu, M.; Wong, A.H.Y.; Schwarz, H. Epstein–Barr virus-encoded LMP1 induces ectopic CD137 expression on Hodgkin and Reed–Sternberg cells via the PI3K-AKT-mTOR pathway. Leuk. Lymphoma 2019, 60, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, L.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2019, 4, e000733. [Google Scholar] [CrossRef]
- Rajendran, S.; Ho, W.T.; Schwarz, H. CD137 signaling in Hodgkin and Reed-Sternberg cell lines induces IL-13 secretion, immune deviation and enhanced growth. OncoImmunology 2016, 5, e1160188. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, C.; Zhang, H.; Duan, Z.; Liu, Q.; Li, J.; Zong, Q.; Yu, W.; Liu, F.; Duan, W.; et al. Characteristics of Immunological Events in Epstein-Barr Virus Infection in Children with Infectious Mononucleosis. Front. Pediatr. 2023, 11, 1060053. [Google Scholar] [CrossRef]
- Eskdale, J.; Kube, D.; Tesch, H.; Gallagher, G. Mapping of the human IL10 gene and further characterization of the 5’ flanking sequence. Immunogenetics 1997, 46, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-Driven LMP1 and IFN-γ Up-Regulate PD-L1 in Nasopharyngeal Carcinoma: Implications for Oncotargeted Therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, W.; Zhang, W.; Wang, W.; Song, Y.; Xiao, H.; Luo, B. Constitutive Activation of the Canonical NF-ΚB Signaling Pathway in EBV-Associated Gastric Carcinoma. Virology 2019, 532, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yang, L.; Xiao, L.; Tang, M.; Liu, L.; Li, Z.; Deng, M.; Sun, L.; Cao, Y. Down-Regulation of EBV-LMP1 Radio-Sensitizes Nasal Pharyngeal Carcinoma Cells via NF-ΚB Regulated ATM Expression. PLoS ONE 2011, 6, e24647. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimičić, P.; Batović, M.; Stojanović Marković, A.; Židovec-Lepej, S. Deciphering the Role of Epstein–Barr Virus Latent Membrane Protein 1 in Immune Modulation: A Multifaced Signalling Perspective. Viruses 2024, 16, 564. https://doi.org/10.3390/v16040564
Šimičić P, Batović M, Stojanović Marković A, Židovec-Lepej S. Deciphering the Role of Epstein–Barr Virus Latent Membrane Protein 1 in Immune Modulation: A Multifaced Signalling Perspective. Viruses. 2024; 16(4):564. https://doi.org/10.3390/v16040564
Chicago/Turabian StyleŠimičić, Petra, Margarita Batović, Anita Stojanović Marković, and Snjezana Židovec-Lepej. 2024. "Deciphering the Role of Epstein–Barr Virus Latent Membrane Protein 1 in Immune Modulation: A Multifaced Signalling Perspective" Viruses 16, no. 4: 564. https://doi.org/10.3390/v16040564