
Evaluation of Bispecific T-Cell Engagers Targeting Murine Cytomegalovirus

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
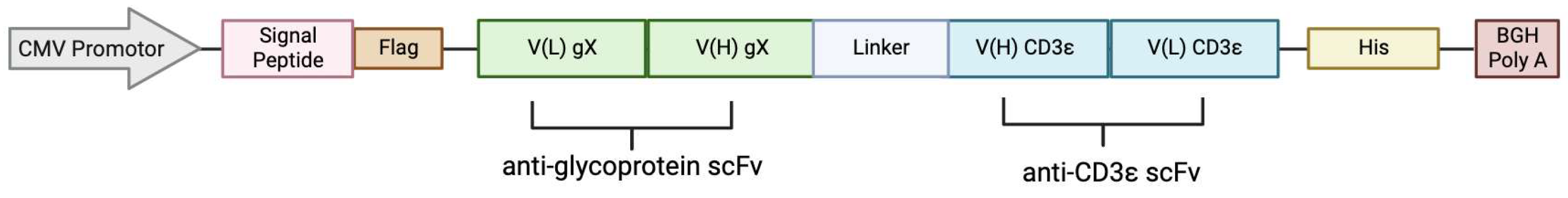
2.1. BiTE Plasmids
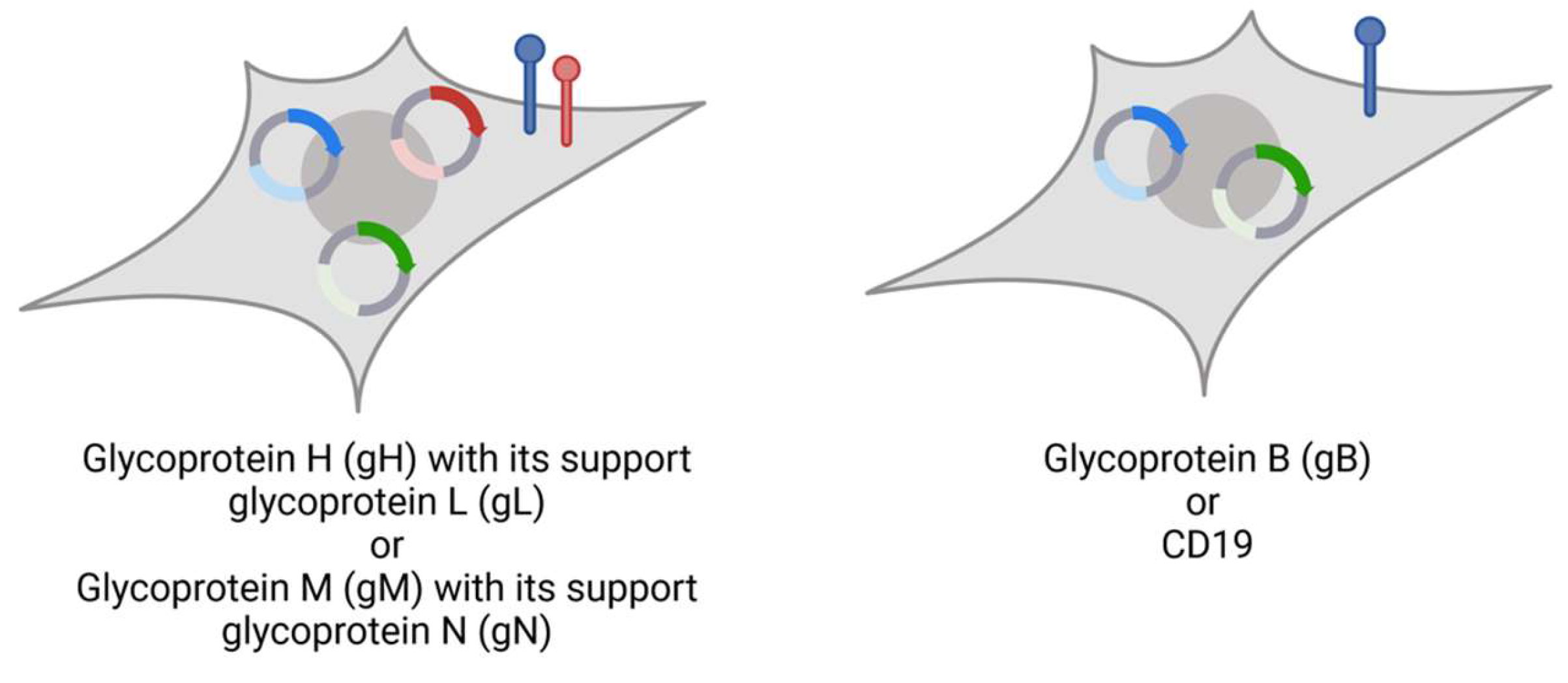
2.2. Plasmids for Generation of Reporter Cells
2.3. Cell Culture
2.3.1. HEK293T Reporter Cell Lines
2.3.2. CB15 T Cells
2.4. Transfection
2.5. Lentivirus Production
2.6. Transduction
2.7. Generation of BiTE
2.8. Flow Cytometry
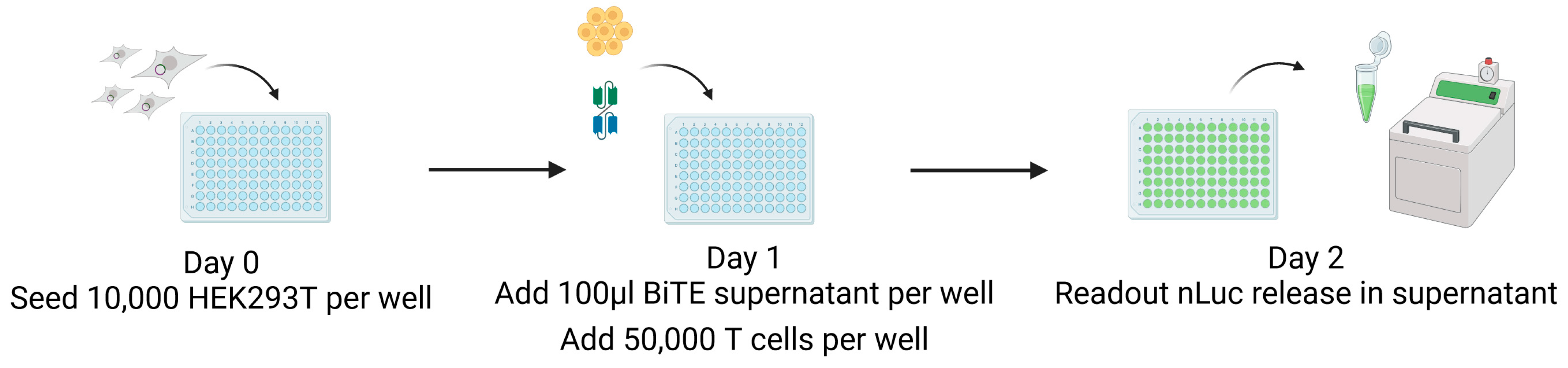
2.9. CB15 T-Cell-Induced Killing: nLuc Assay
3. Results
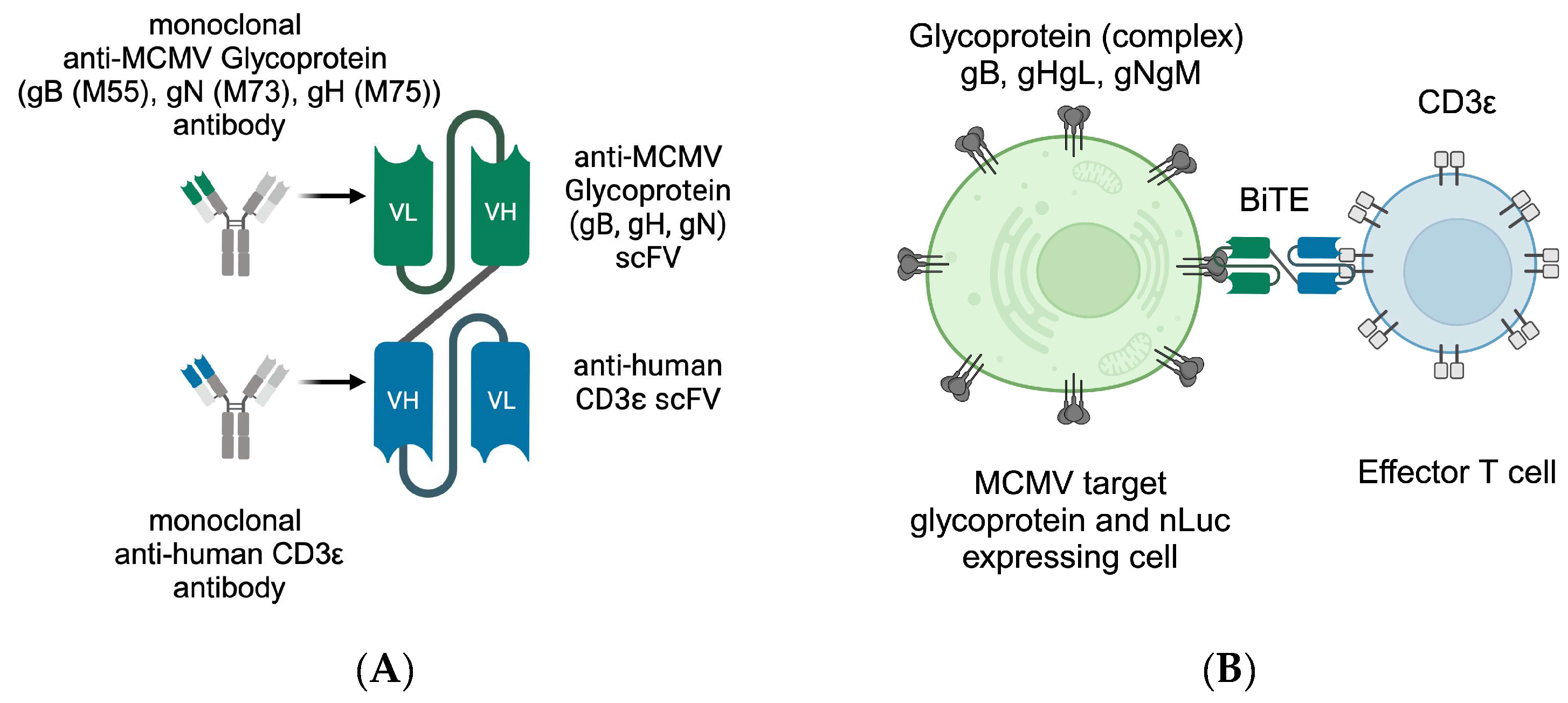
3.1. Design of Bispecific T-Cell Engager (BiTE)
3.2. BiTE Expression and Binding Ability
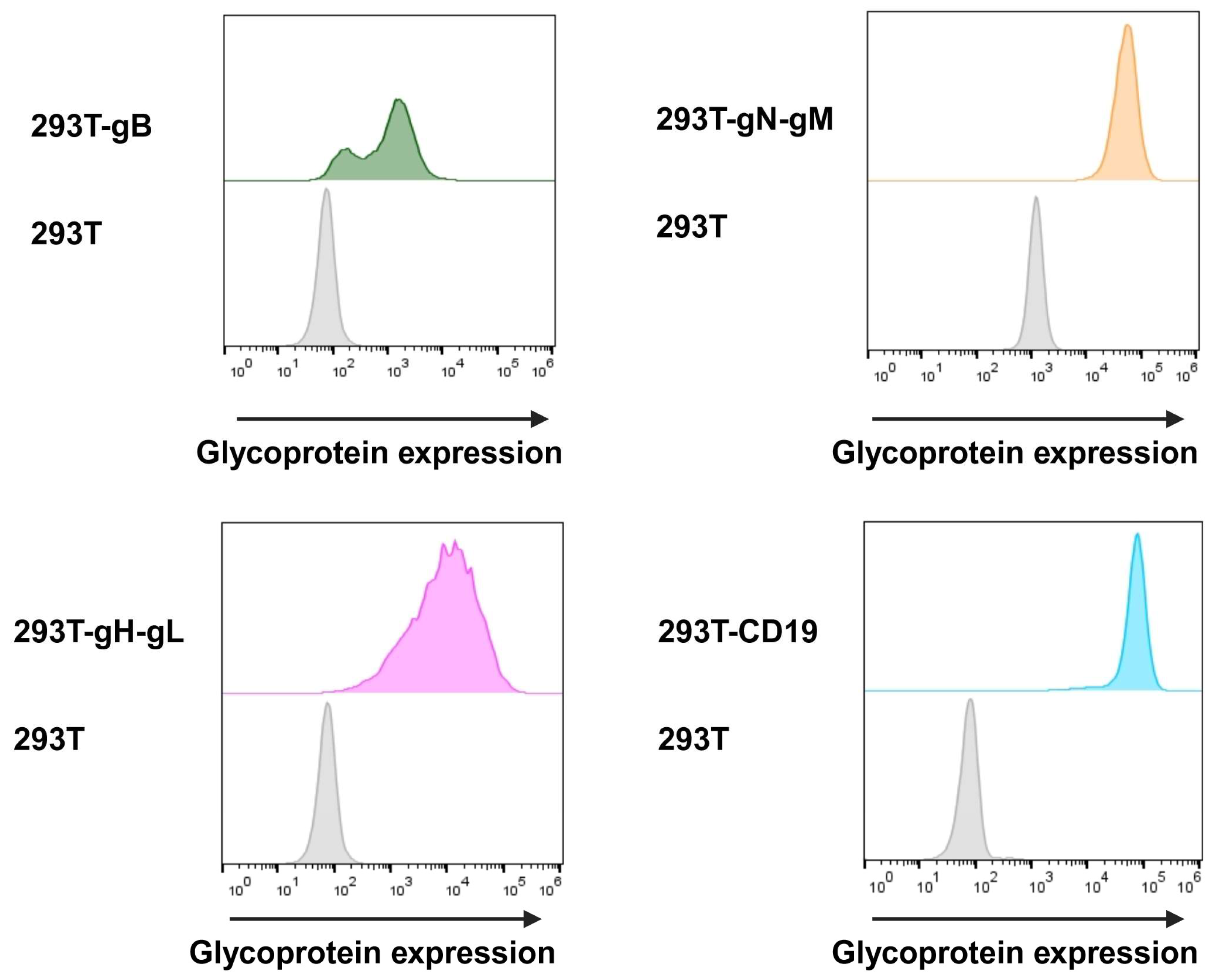
3.2.1. Glycoprotein Expression
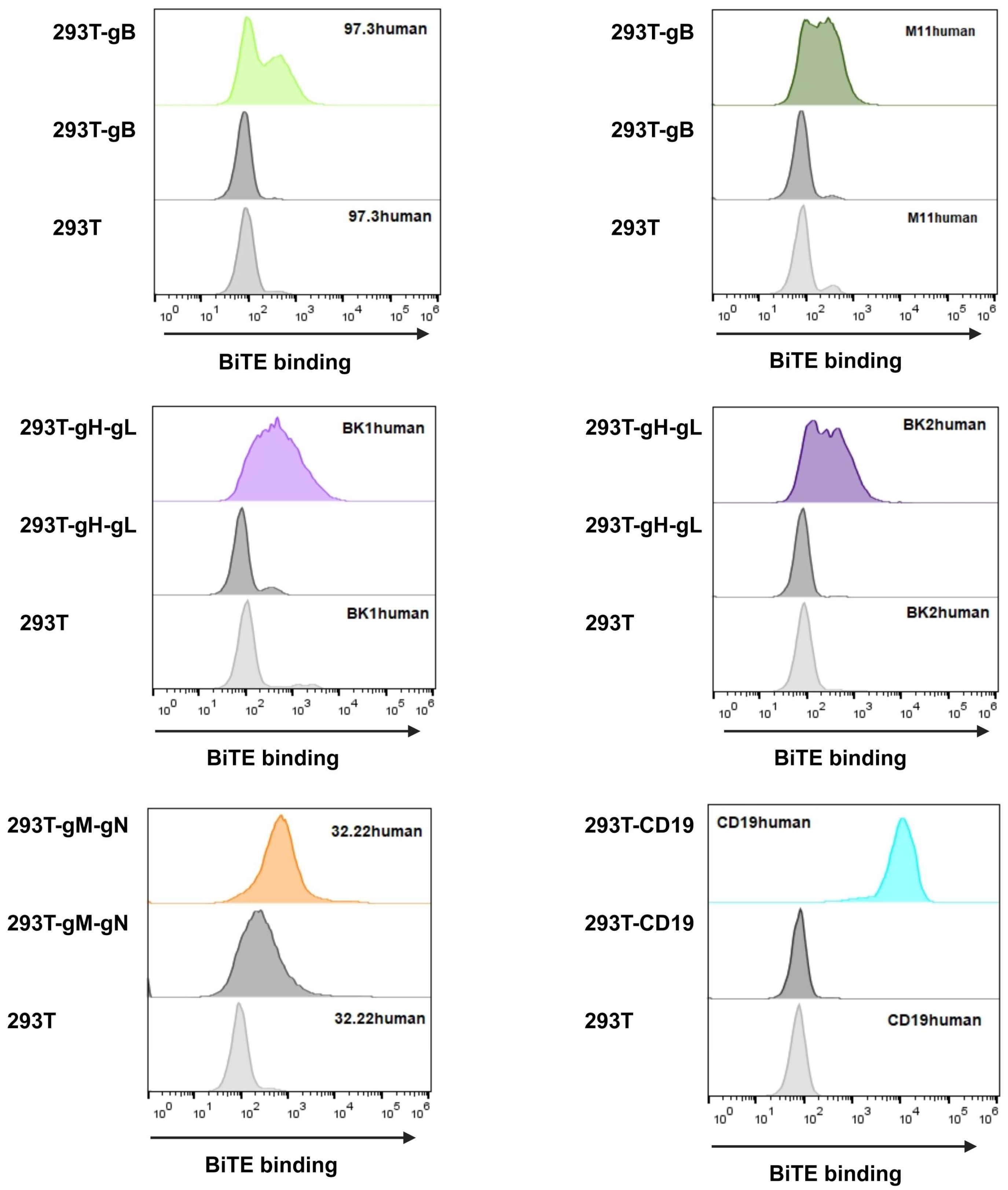
3.2.2. BiTE Binding Ability
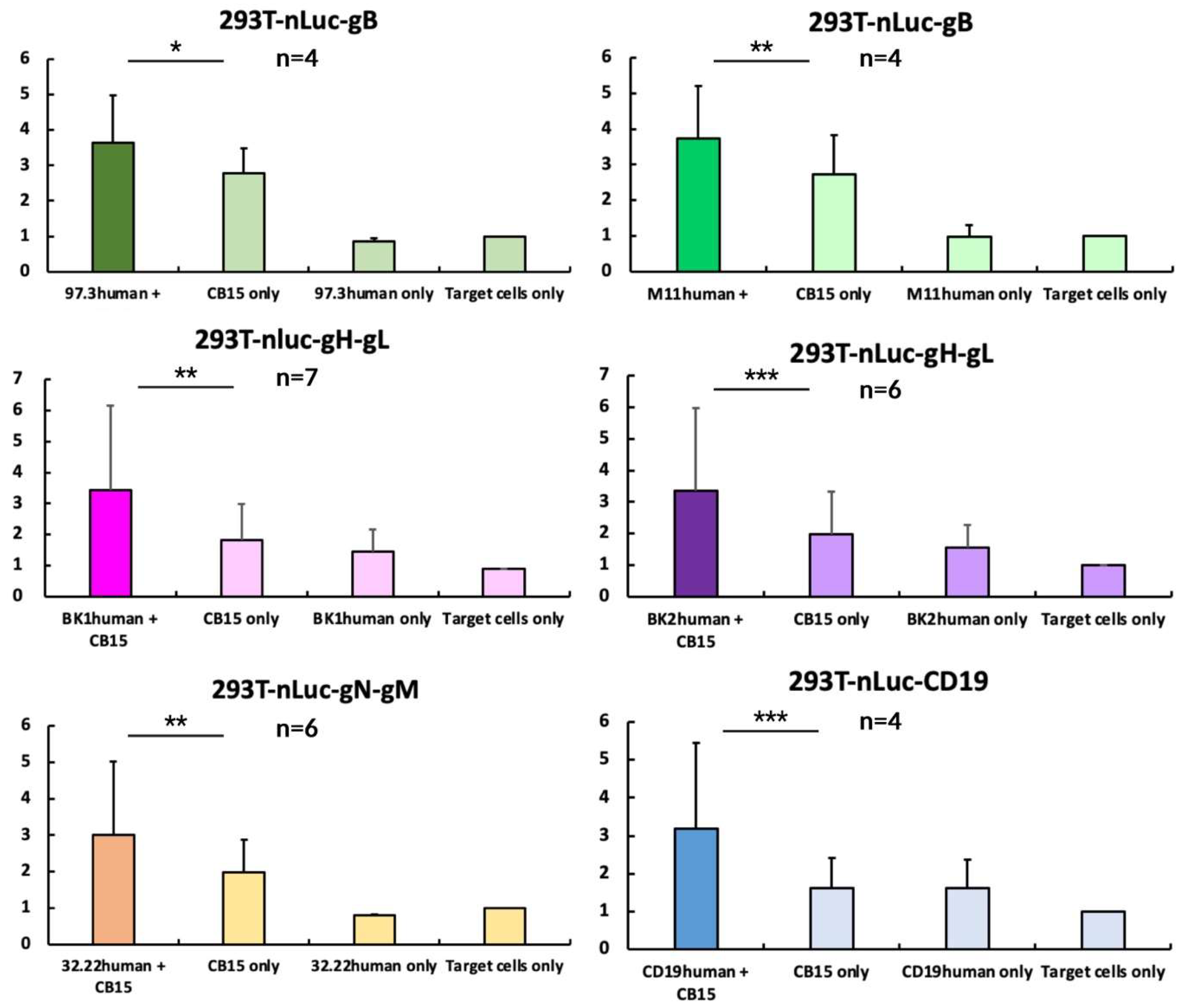
3.3. Design of a Nano-Luciferase-Release Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azevedo, L.S.; Pierrotti, L.C.; Abdala, E.; Costa, S.F.; Strabelli, T.M.V.; Campos, S.V.; Ramos, J.F.; Latif, A.Z.A.; Litvinov, N.; Maluf, N.Z.; et al. Cytomegalovirus infection in transplant recipients. Clinics 2015, 70, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef]
- Rafailidis, P.I.; Mourtzoukou, E.G.; Varbobitis, I.C.; Falagas, M.E. Severe cytomegalovirus infection in apparently immunocompetent patients: A systematic review. Virol. J. 2008, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Eddleston, M.; Peacock, S.; Juniper, M.; Warrell, D.A. Severe cytomegalovirus infection in immunocompetent patients. Clin. Infect. Dis. Off. 1997, 24, 52–56. [Google Scholar] [CrossRef]
- Limaye, A.P.; Raghu, G.; Koelle, D.M.; Ferrenberg, J.; Huang, M.-L.; Boeckh, M. High incidence of ganciclovir-resistant cytomegalovirus infection among lung transplant recipients receiving preemptive therapy. J. Infect. Dis. 2002, 185, 20–27. [Google Scholar] [CrossRef]
- Humar, A.; Lebranchu, Y.; Vincenti, F.; Blumberg, E.A.; Punch, J.D.; Limaye, A.P.; Abramowicz, D.; Jardine, A.G.; Voulgari, A.T.; Ives, J.; et al. The efficacy and safety of 200 days valganciclovir cytomegalovirus prophylaxis in high-risk kidney transplant recipients. Am. J. Transplant. 2010, 10, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.C.; Sia, I.G.; Tau, K.R.; Espy, M.J.; Smith, T.F.; Chou, S.; Paya, C.V. Novel mutation in the CMV UL97 gene associated with resistance to ganciclovir therapy. Transplantation 1999, 67, 755–757. [Google Scholar] [CrossRef]
- Ariza-Heredia, E.J.; Nesher, L.; Chemaly, R.F. Cytomegalovirus diseases after hematopoietic stem cell transplantation: A mini-review. Cancer Lett. 2014, 342, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bowman, L.J.; Melaragno, J.I.; Brennan, D.C. Letermovir for the management of cytomegalovirus infection. Expert Opin. Investig. Drugs 2017, 26, 235–241. [Google Scholar] [CrossRef]
- Perchetti, G.A.; Biernacki, M.A.; Xie, H.; Castor, J.; Joncas-Schronce, L.; Ueda Oshima, M.; Kim, Y.; Jerome, K.R.; Sandmaier, B.M.; Martin, P.J.; et al. Cytomegalovirus breakthrough and resistance during letermovir prophylaxis. Bone Marrow Transplant. 2023, 58, 430–436. [Google Scholar] [CrossRef]
- Balani, S.S.; Sadiq, S.; Jensen, C.J.; Kizilbash, S.J. Prevention and management of CMV infection in pediatric solid organ transplant recipients. Front. Pediatr. 2023, 11, 1098434. [Google Scholar] [CrossRef]
- El Chaer, F.; Shah, D.P.; Chemaly, R.F. How I treat resistant cytomegalovirus infection in hematopoietic cell transplantation recipients. Blood 2016, 128, 2624–2636. [Google Scholar] [CrossRef] [PubMed]
- Holmes-Liew, C.-L.; Holmes, M.; Beagley, L.; Hopkins, P.; Chambers, D.; Smith, C.; Khanna, R. Adoptive T-cell immunotherapy for ganciclovir-resistant CMV disease after lung transplantation. Clin. Transl. Immunol. 2015, 4, e35. [Google Scholar] [CrossRef]
- Limaye, A.P.; Green, M.L.; Edmison, B.C.; Stevens-Ayers, T.; Chatterton-Kirchmeier, S.; Geballe, A.P.; Singh, N.; Boeckh, M. Prospective Assessment of Cytomegalovirus Immunity in High-Risk Donor-Seropositive/Recipient-Seronegative Liver Transplant Recipients Receiving Either Preemptive Therapy or Antiviral Prophylaxis. J. Infect. Dis. 2019, 220, 752–760. [Google Scholar] [CrossRef]
- Green, M.L.; Leisenring, W.M.; Xie, H.; Walter, R.B.; Mielcarek, M.; Sandmaier, B.M.; Riddell, S.R.; Boeckh, M. CMV reactivation after allogeneic HCT and relapse risk: Evidence for early protection in acute myeloid leukemia. Blood 2013, 122, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Boeckh, M.; Geballe, A.P. Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Investig. 2011, 121, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Teira, P.; Battiwalla, M.; Ramanathan, M.; Barrett, A.J.; Ahn, K.W.; Chen, M.; Green, J.S.; Saad, A.; Antin, J.H.; Savani, B.N.; et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: A CIBMTR analysis. Blood 2016, 127, 2427–2438. [Google Scholar] [CrossRef]
- Benn, H.; Rowley, S.D. Chapter 58—Bone Marrow and Peripheral Blood Stem Cell Transplantation. In Blood Banking and Transfusion Medicine, 2nd ed.; Hillyer, C.D., Silberstein, L.E., Ness, P.M., Anderson, K.C., Roback, J.D., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2007; pp. 787–822. Available online: https://www.sciencedirect.com/science/article/pii/B9780443069819500636 (accessed on 3 May 2024).
- Dunn, H.S.; Haney, D.J.; Ghanekar, S.A.; Stepick-Biek, P.; Lewis, D.B.; Maecker, H.T. Dynamics of CD4 and CD8 T Cell Responses to Cytomegalovirus in Healthy Human Donors. J. Infect. Dis. 2002, 186, 15–22. [Google Scholar] [CrossRef]
- Lim, E.Y.; Jackson, S.E.; Wills, M.R. The CD4+ T Cell Response to Human Cytomegalovirus in Healthy and Immunocompromised People. Front. Cell. Infect. Microbiol. 2020, 10, 202. [Google Scholar] [CrossRef]
- Verma, S.; Weiskopf, D.; Gupta, A.; McDonald, B.; Peters, B.; Sette, A.; Benedict, C.A. Cytomegalovirus-Specific CD4 T Cells Are Cytolytic and Mediate Vaccine Protection. J. Virol. 2016, 90, 650–658. [Google Scholar] [CrossRef]
- Zangger, N.; Oxenius, A. T cell immunity to cytomegalovirus infection. Curr. Opin. Immunol. 2022, 77, 102185. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Mutter, W.; Münch, K.; Bühring, H.J.; Koszinowski, U.H. CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. J. Virol. 1987, 61, 3102–3108. [Google Scholar] [CrossRef]
- Koszinowski, U.H.; Reddehase, M.J.; Jonjic, S. The role of CD4 and CD8 T cells in viral infections. Curr. Opin. Immunol. 1991, 3, 471–475. [Google Scholar] [CrossRef]
- Holtappels, R.; Böhm, V.; Podlech, J.; Reddehase, M. CD8 T-cell-based immunotherapy of cytomegalovirus infection: “Proof of concept” provided by the murine model. Med. Microbiol. Immunol. 2008, 197, 125–134. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gökbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete Hematologic and Molecular Response in Adult Patients With Relapsed/Refractory Philadelphia Chromosome–Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment With Blinatumomab: Results From a Phase II, Single-Arm, Multicenter Study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [Google Scholar] [CrossRef]
- Fisher, M.A.; Lloyd, M.L. A Review of Murine Cytomegalovirus as a Model for Human Cytomegalovirus Disease—Do Mice Lie? Int. J. Mol. Sci. 2020, 22, 214. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Lemmermann, N.A.W. Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”. Viruses 2018, 10, 693. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar] [CrossRef] [PubMed]
- Cordsmeier, A.; Bednar, C.; Kübel, S.; Bauer, L.; Ensser, A. Re-Analysis of the Widely Used Recombinant Murine Cytomegalovirus MCMV-m157luc Derived from the Bacmid pSM3fr Confirms Its Hybrid Nature. Int. J. Mol. Sci. 2023, 24, 14102. [Google Scholar] [CrossRef]
- Brey, C.U.; Proff, J.; Teufert, N.; Salzer, B.; Brozy, J.; Münz, M.; Pendzialek, J.; Ensser, A.; Holter, W.; Lehner, M. A gB/CD3 bispecific BiTE antibody construct for targeting Human Cytomegalovirus-infected cells. Sci. Rep. 2018, 8, 17453. [Google Scholar] [CrossRef]
- Jackson, S.E.; Mason, G.M.; Wills, M.R. Human cytomegalovirus immunity and immune evasion. Virus Res. 2011, 157, 151–160. [Google Scholar] [CrossRef]
- Wu, Z.; Lau, C.M.; Sottile, R.; Le Luduec, J.-B.; Panjwani, M.K.; Conaty, P.M.; Srpan, K.; Laib Sampaio, K.; Mertens, T.; Adler, S.P.; et al. Human Cytomegalovirus Infection Promotes Expansion of a Functionally Superior Cytoplasmic CD3+ NK Cell Subset with a Bcl11b-Regulated T Cell Signature. J. Immunol. 2021, 207, 2534–2544. [Google Scholar] [CrossRef]
- Wu, Z.; Frascaroli, G.; Bayer, C.; Schmal, T.; Mertens, T. Interleukin-2 from Adaptive T Cells Enhances Natural Killer Cell Activity against Human Cytomegalovirus-Infected Macrophages. J. Virol. 2015, 89, 6435–6441. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.H.; Guseva, N.V.; Ball, B.L.; Heusel, J.W. Characterization of Murine Cytomegalovirus m157 from Infected Cells and Identification of Critical Residues Mediating Recognition by the NK Cell Receptor, Ly49H. J. Immunol. 2008, 181, 265–275. [Google Scholar] [CrossRef]
- Smith, H.R.C.; Heusel, J.W.; Mehta, I.K.; Kim, S.; Dorner, B.G.; Naidenko, O.V.; Iizuka, K.; Furukawa, H.; Beckman, D.L.; Pingel, J.T.; et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 8826–8831. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, M.K.; Compton, T. Human Cytomegalovirus Glycoprotein B Is Required for Virus Entry and Cell-to-Cell Spread but Not for Virion Attachment, Assembly, or Egress. J. Virol. 2009, 83, 3891–3903. [Google Scholar] [CrossRef]
- Burke, H.G.; Heldwein, E.E. Crystal Structure of the Human Cytomegalovirus Glycoprotein, B. PLoS Pathog. 2015, 11, e1005227. [Google Scholar] [CrossRef]
- Meng, W.; Tang, A.; Ye, X.; Gui, X.; Li, L.; Fan, X.; Schultz, R.D.; Freed, D.C.; Ha, S.; Wang, D.; et al. Targeting Human-Cytomegalovirus-Infected Cells by Redirecting T Cells Using an Anti-CD3/Anti-Glycoprotein B Bispecific Antibody. Antimicrob. Agents Chemother. 2018, 62, e01719-17. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Mach, M.; Britt, W.J. Human Cytomegalovirus Infection Elicits a Glycoprotein M (gM)/gN-Specific Virus-Neutralizing Antibody Response. J. Virol. 2006, 80, 4591–4600. [Google Scholar] [CrossRef] [PubMed]
- Lilleri, D.; Kabanova, A.; Lanzavecchia, A.; Gerna, G. Antibodies against neutralization epitopes of human cytomegalovirus gH/gL/pUL128-130-131 complex and virus spreading may correlate with virus control in vivo. J. Clin. Immunol. 2012, 32, 1324–1331. [Google Scholar] [CrossRef]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Chou, S.; Gojobori, T.; Hanada, K.; Guo, J.J.; Rawlinson, W.; Britt, W.; Mach, M.; et al. Human cytomegalovirus glycoprotein N (gpUL73-gN) genomic variants: Identification of a novel subgroup, geographical distribution and evidence of positive selective pressure. J. Gen. Virol. 2003, 84, 647–655. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Paša-Tolić, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S.; et al. Identification of Proteins in Human Cytomegalovirus (HCMV) Particles: The HCMV Proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Ye, X.; Freed, D.C.; Li, L.; Ku, Z.; Xiong, W.; Gao, P.; Liu, X.; Montgomery, D.; Xu, W.; et al. Potent Bispecific Neutralizing Antibody Targeting Glycoprotein B and the gH/gL/pUL128/130/131 Complex of Human Cytomegalovirus. Antimicrob. Agents Chemother. 2021, 65, e02422-20. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Zhu, M.; Wu, B.; Brandl, C.; Johnson, J.; Wolf, A.; Chow, A.; Doshi, S. Blinatumomab, a Bispecific T-cell Engager (BiTE®) for CD-19 Targeted Cancer Immunotherapy: Clinical Pharmacology and Its Implications. Clin. Pharmacokinet. 2016, 55, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Bednar, C. The Basis of Chimeric Antigen Receptor-Based Adoptive T Cell Immunotherapy in the Murine Cytomegalovirus Infection Model; Friedrich-Alexander-Universität Erlangen-Nürnberg: Erlangen, Germany, 2024. [Google Scholar]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef]
- Biesinger, B.; Muller-Fleckenstein, I.; Simmer, B.; Lang, G.; Wittmann, S.; Platzer, E.; Desrosiers, R.C.; Fleckenstein, B. Stable Growth Transformation of Human T Lymphocytes by Herpesvirus saimiri. Proc. Natl. Acad. Sci. USA 1992, 89, 3116–3119. [Google Scholar] [CrossRef] [PubMed]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Mach, M.; Kropff, B.; Kryzaniak, M.; Britt, W. Complex formation by glycoproteins M and N of human cytomegalovirus: Structural and functional aspects. J. Virol. 2005, 79, 2160–2170. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.; Hernandez, R.; Noriega, V.; Tortorella, D. Human cytomegalovirus gH stability and trafficking are regulated by ER-associated degradation and transmembrane architecture. Sci. Rep. 2016, 6, 23692. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.; Gopalakrishnan, R.; Choi, S.; Prakash, R.; Natarajan, V.; Prins, R.; Gong, S.; Chitnis, S.D.; Kahn, M.; Han, X.; et al. Development and characterization of a novel luciferase based cytotoxicity assay. Sci. Rep. 2018, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.P.; Valentine, M.C.; Gao, J.; Pingel, J.T.; Yokoyama, W.M. Stability of murine cytomegalovirus genome after in vitro and in vivo passage. J. Virol. 2010, 84, 2623–2628. [Google Scholar] [CrossRef]
- Van Damme, E.; Van Loock, M. Functional annotation of human cytomegalovirus gene products: An update. Front. Microbiol. 2014, 5, 218. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lorenczewski, G.; Bailis, J.M.; Stienen, S.; Friedrich, M.; Lee, F.; van der Vegt, B.; de Vries, E.G.E.; de Groot, D.J.A.; Lub-de Hooge, M.N. Mesothelin/CD3 Half-Life–Extended Bispecific T-Cell Engager Molecule Shows Specific Tumor Uptake and Distributes to Mesothelin and CD3-Expressing Tissues. J. Nucl. Med. 2021, 62, 1797–1804. [Google Scholar] [CrossRef]
- Proff, J.; Walterskirchen, C.; Brey, C.; Geyeregger, R.; Full, F.; Ensser, A.; Lehner, M.; Holter, W. Cytomegalovirus-Infected Cells Resist T Cell Mediated Killing in an HLA-Recognition Independent Manner. Front. Microbiol. 2016, 7, 844. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Zheng, P.-P.; Kros, J.M.; Wang, G. Elusive Neurotoxicity in T Cell-Boosting Anticancer Therapies. Trends Immunol. 2019, 40, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.S.; Schiller, G.; Benjamin, R.; Jia, C.; Zhang, A.; Zhu, M.; Zimmerman, Z.; Topp, M.S. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: Management and mitigating factors. Ann. Hematol. 2019, 98, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Bednar, C.; Ensser, A. CARs-A New Perspective to HCMV Treatment. Viruses 2021, 13, 1563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Penny, H.L.; Kroenke, M.A.; Bautista, B.; Hainline, K.; Chea, L.S.; Parnes, J.; Mytych, D.T. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J. Immunother. Cancer 2022, 10, e004225. [Google Scholar] [CrossRef]
- Penny, H.L.; Hainline, K.; Theoharis, N.; Wu, B.; Brandl, C.; Webhofer, C.; McComb, M.; Wittemer-Rump, S.; Koca, G.; Stienen, S.; et al. Characterization and root cause analysis of immunogenicity to pasotuxizumab (AMG 212), a prostate-specific membrane antigen-targeting bispecific T-cell engager therapy. Front. Immunol. 2023, 14, 1261070. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Hybridoma | Plasmid | Encoded BiTE |
|---|---|---|---|
| gB | 97.3 | pCDNA-97.3huCD3 | 97.3human |
| M11 | pCDNA-M11huCD3 | M11human | |
| gN | 32.22 | pCDNA-32.22huCD3 | 32.22human |
| gH | BK1 | pCDNA-BK1huCD3 | BK1human |
| BK2 | pCDNA-BK2huCD3 | BK2human | |
| CD19 | HD37 | pCDNA-CD19huCD3 | CD19human |
| Plasmid | Resistance Marker |
|---|---|
| pLV-gB | Puromycin |
| pLV-gN | Puromycin |
| pLV-gM | Blasticidin |
| pLV-gH | Puromycin |
| pLV-gL | Blasticidin |
| pLV-CD19 | Puromycin |
| pLV-nLuc | Hygromycin B |
| Antibody | Origin | Dilution | Assay |
|---|---|---|---|
| Anti-Flag® BioM2 | Mouse, #F9291, Merck | 1:1000 | BiTE detection gM detection |
| Anti-HA | Mouse, #901515, Biolegend (San Diego, CA, USA) | 1:1000 | gN detection |
| BK2 (Hybridoma) | Michael Mach, Virology Erlangen | 100 μL hybridoma supernatant | gH detection |
| M11 (Hybridoma) | Michael Mach, Virology Erlangen | 100 μL hybridoma supernatant | gB detection |
| anti-CD19-647 | BioLegend, #363040 | 1:200 | CD19 detection |
| anti-mouse IgG-647 | Thermo Fisher Scientific, #A31571 | 1:1000 | Secondary antibody |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menschikowski, H.; Bednar, C.; Kübel, S.; Hermann, M.; Bauer, L.; Thomas, M.; Cordsmeier, A.; Ensser, A. Evaluation of Bispecific T-Cell Engagers Targeting Murine Cytomegalovirus. Viruses 2024, 16, 869. https://doi.org/10.3390/v16060869
Menschikowski H, Bednar C, Kübel S, Hermann M, Bauer L, Thomas M, Cordsmeier A, Ensser A. Evaluation of Bispecific T-Cell Engagers Targeting Murine Cytomegalovirus. Viruses. 2024; 16(6):869. https://doi.org/10.3390/v16060869
Chicago/Turabian StyleMenschikowski, Hanna, Christopher Bednar, Sabrina Kübel, Manuel Hermann, Larissa Bauer, Marco Thomas, Arne Cordsmeier, and Armin Ensser. 2024. "Evaluation of Bispecific T-Cell Engagers Targeting Murine Cytomegalovirus" Viruses 16, no. 6: 869. https://doi.org/10.3390/v16060869