
HIV-1 Tat-Mediated Human Müller Glial Cell Senescence Involves Endoplasmic Reticulum Stress and Dysregulated Autophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Treatments
2.3. Western Blotting
2.4. LC3 Accumulation and p62 Degradation Assays
2.5. Immunocytochemistry
2.6. Evaluation of Autophagosome Formation and Maturation
2.7. Senescence-Associated (SA) β-Galactosidase (Gal) Staining Assay
2.8. Statistical Analysis
3. Results
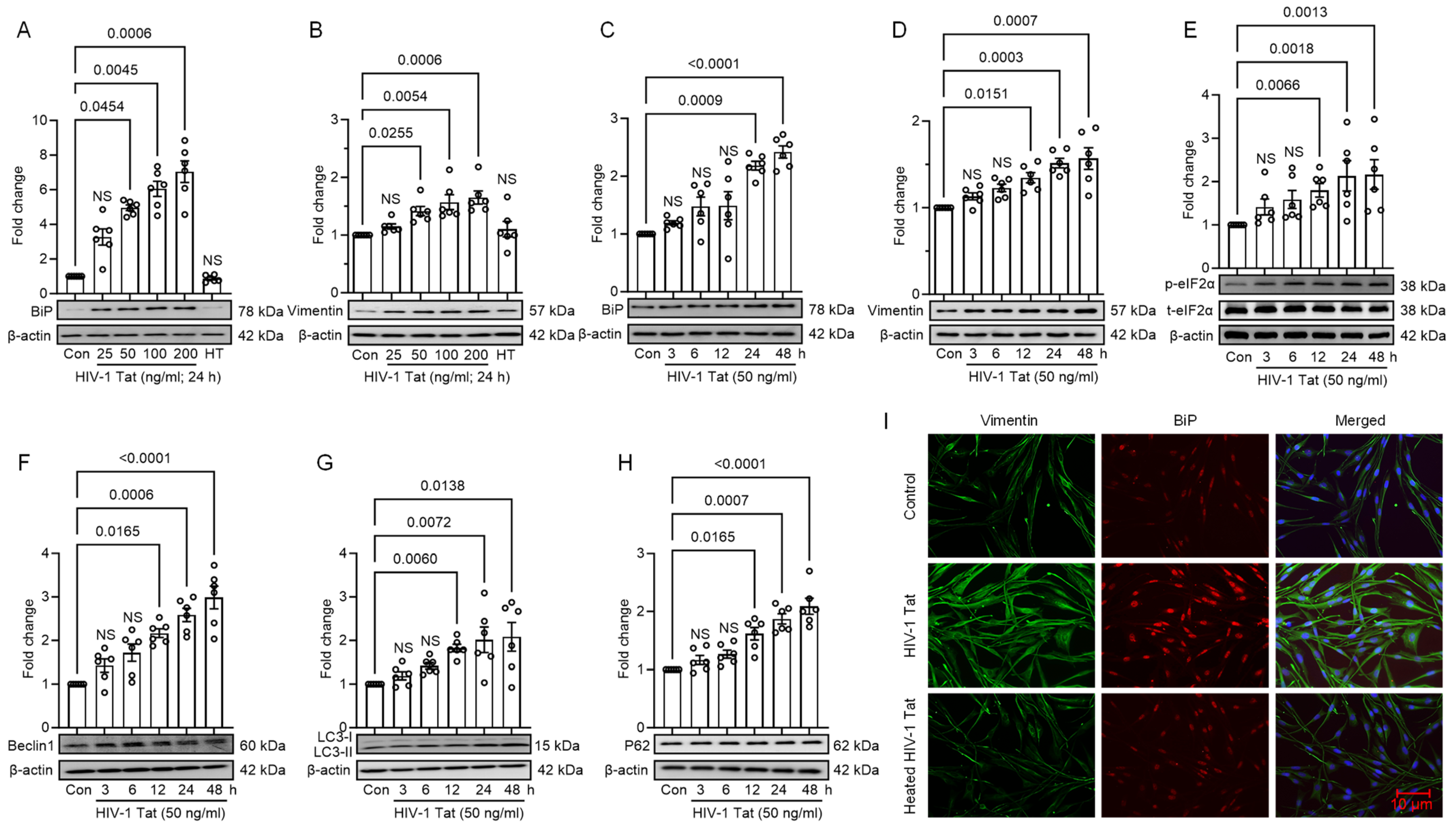
3.1. HIV-1 Tat Induces ER Stress and Autophagy in MIO-M1 Cells
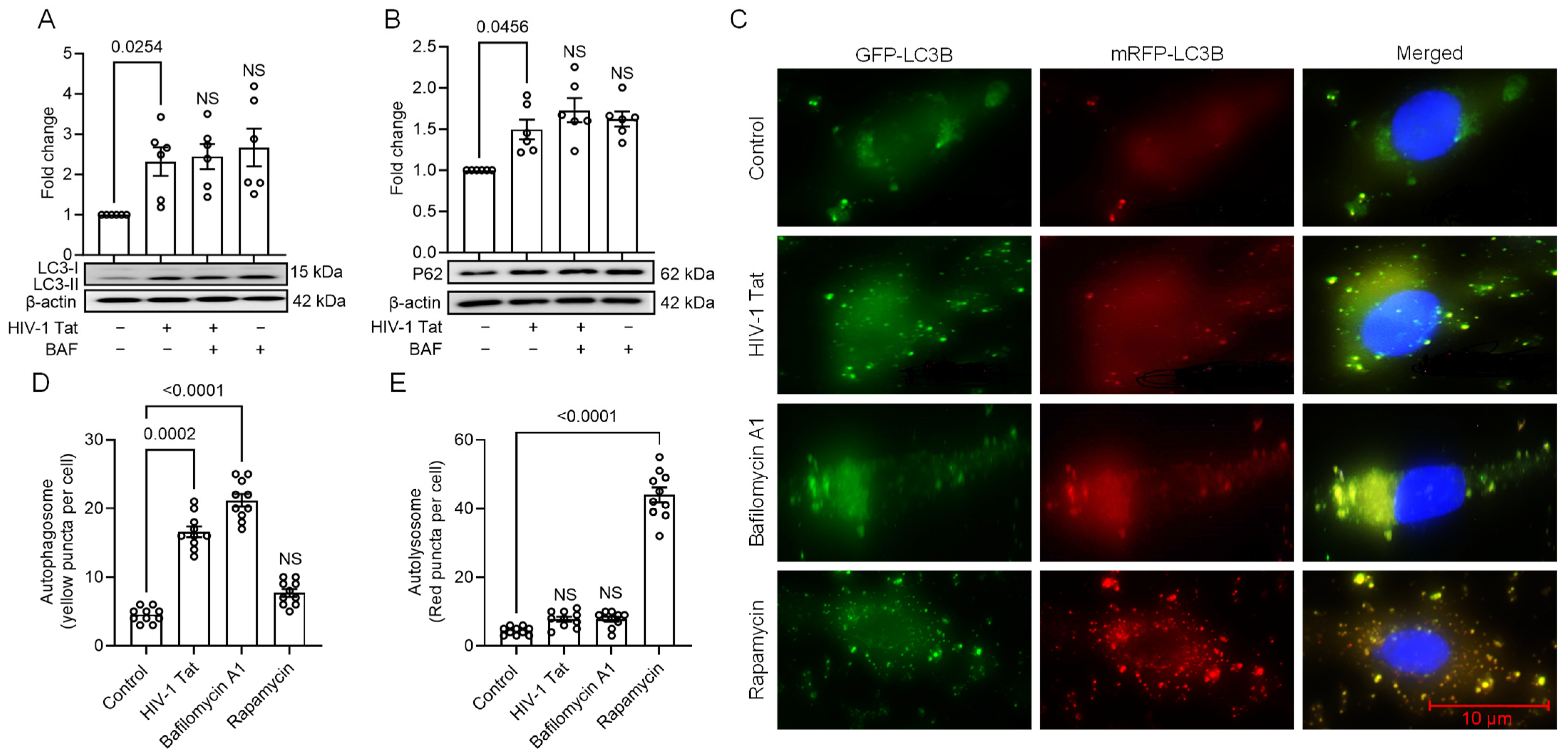
3.2. HIV-1 Tat-Mediated Activation of ER Stress Results in Dysregulated Autophagy in MIO-M1 Cells
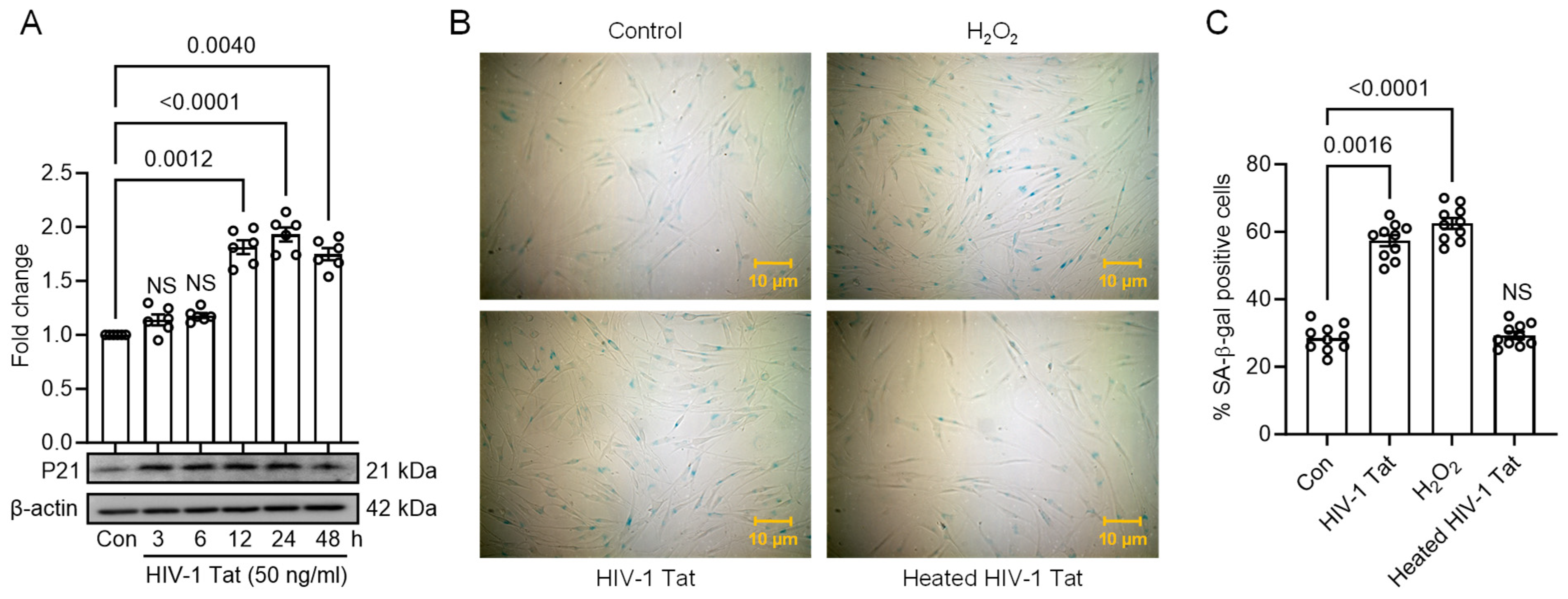
3.3. HIV-1 Tat Activates ER Stress, Autophagy, and Senescence in MIO-M1 Cells
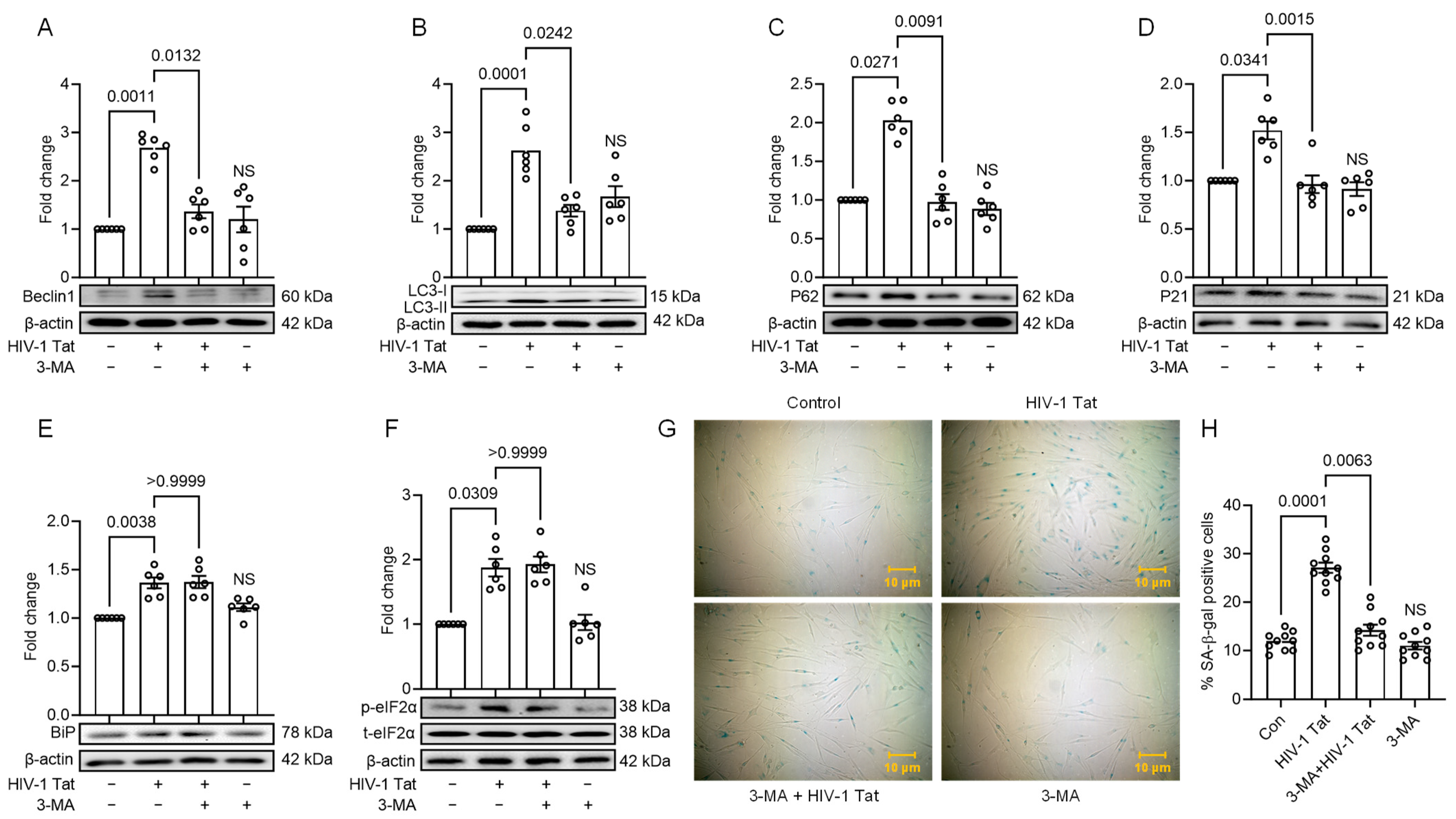
3.4. HIV-1 Tat-Mediated Senescence Signalling Involves Upstream Activation of ER Stress
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feroze, K.B.; Wang, J. Ocular Manifestations of HIV. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Luo, J.; Jing, D.; Kozak, I.; Huiming, Z.; Siying, C.; Yezhen, Y.; Xin, Q.; Luosheng, T.; Adelman, R.A.; Forster, S.H. Prevalence of ocular manifestations of HIV/AIDS in the highly active antiretroviral therapy (HAART) era: A different spectrum in Central South China. Ophthalmic Epidemiol. 2013, 20, 170–175. [Google Scholar] [CrossRef]
- Zhang, K.; Yang, C.; Yao, H.; Lin, A. [3 + 2] Cycloaddition Reaction of in Situ Formed Azaoxyallyl Cations with Aldehydes: An Approach to Oxazolidin-4-ones. Org. Lett. 2016, 18, 4618–4621. [Google Scholar] [CrossRef]
- Moraes, H.V., Jr. Ocular manifestations of HIV/AIDS. Curr. Opin. Ophthalmol. 2002, 13, 397–403. [Google Scholar] [CrossRef]
- Cunningham, E.T., Jr.; Margolis, T.P. Ocular manifestations of HIV infection. N. Engl. J. Med. 1998, 339, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.R.; Ross, M.L.; Whitcup, S.M. Ocular manifestations of HIV infection. Curr. Opin. Ophthalmol. 1999, 10, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Yeo, T.H.; Yeo, T.K.; Wong, E.P.; Agrawal, R.; Teoh, S.C. Immune recovery uveitis in HIV patients with cytomegalovirus retinitis in the era of HAART therapy-a 5-year study from Singapore. J. Ophthalmic Inflamm. Infect. 2016, 6, 41. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Pathai, S.; Shiels, P.G.; Weiss, H.A.; Gilbert, C.E.; Peto, T.; Bekker, L.G.; Wood, R.; Wong, T.Y.; Lawn, S.D. Ocular parameters of biological ageing in HIV-infected individuals in South Africa: Relationship with chronological age and systemic biomarkers of ageing. Mech. Ageing Dev. 2013, 134, 400–406. [Google Scholar] [CrossRef]
- Deeks, S.G. Immune dysfunction, inflammation, and accelerated aging in patients on antiretroviral therapy. Top. HIV Med. 2009, 17, 118–123. [Google Scholar]
- Jabs, D.A.; Van Natta, M.L.; Sezgin, E.; Pak, J.W.; Danis, R.; Studies of the Ocular Complications of AIDS Research Group. Prevalence of intermediate-stage age-related macular degeneration in patients with acquired immunodeficiency syndrome. Am. J. Ophthalmol. 2015, 159, 1115–1122.e1111. [Google Scholar] [CrossRef]
- Bringmann, A.; Wiedemann, P. Muller glial cells in retinal disease. Ophthalmologica 2012, 227, 1–19. [Google Scholar] [CrossRef]
- Navneet, S.; Wilson, K.; Rohrer, B. Muller Glial Cells in the Macula: Their Activation and Cell-Cell Interactions in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2024, 65, 42. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Wu, Y.; Shao, C.; Qiu, Q. The Role of Muller Cells in Diabetic Macular Edema. Investig. Ophthalmol. Vis. Sci. 2023, 64, 8. [Google Scholar] [CrossRef] [PubMed]
- Abcouwer, S.F. Muller Cell-Microglia Cross Talk Drives Neuroinflammation in Diabetic Retinopathy. Diabetes 2017, 66, 261–263. [Google Scholar] [CrossRef]
- Goldberg, D.E.; Smithen, L.M.; Angelilli, A.; Freeman, W.R. HIV-associated retinopathy in the HAART era. Retina 2005, 25, 633–649; quiz 682–633. [Google Scholar] [CrossRef] [PubMed]
- Nooka, S.; Ghorpade, A. HIV-1-associated inflammation and antiretroviral therapy regulate astrocyte endoplasmic reticulum stress responses. Cell Death Discov. 2017, 3, 17061. [Google Scholar] [CrossRef]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Moon, S.; Jung, H.S. Endoplasmic Reticulum Stress and Dysregulated Autophagy in Human Pancreatic Beta Cells. Diabetes Metab. J. 2022, 46, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Andras, I.E.; Toborek, M. Amyloid beta accumulation in HIV-1-infected brain: The role of the blood brain barrier. IUBMB Life 2013, 65, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.; Sil, S.; Oladapo, A.; Thangaraj, A.; Periyasamy, P.; Buch, S. HIV-1 Tat-mediated microglial ferroptosis involves the miR-204-ACSL4 signaling axis. Redox Biol. 2023, 62, 102689. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Guo, M.L.; Buch, S. Cocaine induces astrocytosis through ER stress-mediated activation of autophagy. Autophagy 2016, 12, 1310–1329. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.L.; Buch, S. Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol. Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFkappaB signaling axis. Brain Behav. Immun. 2019, 80, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Thangaraj, A.; Guo, M.L.; Hu, G.; Callen, S.; Buch, S. Epigenetic Promoter DNA Methylation of miR-124 Promotes HIV-1 Tat-Mediated Microglial Activation via MECP2-STAT3 Axis. J. Neurosci. 2018, 38, 5367–5383. [Google Scholar] [CrossRef]
- Periyasamy, P.; Thangaraj, A.; Kannan, M.; Oladapo, A.; Buch, S. The Epigenetic Role of miR-124 in HIV-1 Tat- and Cocaine-Mediated Microglial Activation. Int. J. Mol. Sci. 2022, 23, 15017. [Google Scholar] [CrossRef]
- Singh, S.; Thangaraj, A.; Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Buch, S. Role of Dysregulated Autophagy in HIV Tat, Cocaine, and cART Mediated NLRP3 Activation in Microglia. J. Neuroimmune Pharmacol. 2023, 18, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Pillai, P.P.; Kannan, M.; Sil, S.; Singh, S.; Thangaraj, A.; Chivero, E.T.; Dagur, R.S.; Tripathi, A.; Hu, G.; Periyasamy, P.; et al. Involvement of lncRNA TUG1 in HIV-1 Tat-Induced Astrocyte Senescence. Int. J. Mol. Sci. 2023, 24, 4330. [Google Scholar] [CrossRef]
- Shmakova, A.; Tsimailo, I.; Kozhevnikova, Y.; Gerard, L.; Boutboul, D.; Oksenhendler, E.; Tuaillon, E.; Rivault, A.; Germini, D.; Vassetzky, Y.; et al. HIV-1 Tat is present in the serum of people living with HIV-1 despite viral suppression. Int. J. Infect. Dis. 2024, 142, 106994. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.B.; Santamaria, U.A.; Bachani, M.; Demarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33 (Suppl. 2), S145–S157. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Neuveut, C.; Tiffany, H.L.; Benkirane, M.; Rich, E.A.; Murphy, P.M.; Jeang, K.T. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. USA 2000, 97, 11466–11471. [Google Scholar] [CrossRef] [PubMed]
- Rayne, F.; Debaisieux, S.; Bonhoure, A.; Beaumelle, B. HIV-1 Tat is unconventionally secreted through the plasma membrane. Cell Biol. Int. 2010, 34, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Hennig, B.; Toborek, M. Intact lipid rafts regulate HIV-1 Tat protein-induced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J. Cereb. Blood Flow. Metab. 2010, 30, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Carosio, R.; Fenoglio, D.; Brenci, S.; Murdaca, G.; Setti, M.; Indiveri, F.; Scabini, S.; Ferrero, E.; Zocchi, M.R. Migration of V delta 1 and V delta 2 T cells in response to CXCR3 and CXCR4 ligands in healthy donors and HIV-1-infected patients: Competition by HIV-1 Tat. Blood 2004, 103, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Thayer, S.A. HIV Tat Protein Selectively Impairs CB(1) Receptor-Mediated Presynaptic Inhibition at Excitatory But Not Inhibitory Synapses. eNeuro 2020, 7, ENEURO.0119-20.2020. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2018, 15, 97–108. [Google Scholar] [CrossRef]
- Reeder, J.E.; Kwak, Y.T.; McNamara, R.P.; Forst, C.V.; D’Orso, I. HIV Tat controls RNA Polymerase II and the epigenetic landscape to transcriptionally reprogram target immune cells. eLife 2015, 4, e08955. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, D.H.; Ganesh, K.N.; Mitra, D. HIV-1 Tat directly binds to NFkappaB enhancer sequence: Role in viral and cellular gene expression. Nucleic Acids Res. 2004, 32, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Callen, S.; Seigel, G.M.; Buch, S.J. HIV-1 Tat-mediated neurotoxicity in retinal cells. J. Neuroimmune Pharmacol. 2011, 6, 399–408. [Google Scholar] [CrossRef]
- Madigan, M.C.; Sadun, A.A.; Rao, N.S.; Dugel, P.U.; Tenhula, W.N.; Gill, P.S. Tumor necrosis factor-alpha (TNF-alpha)-induced optic neuropathy in rabbits. Neurol. Res. 1996, 18, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.D.; Song, M.K.; Smith, I.L.; Karavellas, M.P.; Bartsch, D.U.; Torriani, F.J.; Garcia, C.R.; Freeman, W.R. Intraocular viral and immune pathogenesis of immune recovery uveitis in patients with healed cytomegalovirus retinitis. Retina 2006, 26, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Woodman, S.E.; Benveniste, E.N.; Nath, A.; Berman, J.W. Human immunodeficiency virus type 1 TAT protein induces adhesion molecule expression in astrocytes. J. Neurovirol. 1999, 5, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.; Liu, J.; Nath, A.; Jones, M.; Raghavan, R.; Narayan, O.; Male, D.; Everall, I. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J. Neurovirol. 2000, 6, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.O.; Liu, Y.; Ruan, Y.; Xu, Z.C.; Schantz, L.; He, J.J. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am. J. Pathol. 2003, 162, 1693–1707. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef] [PubMed]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.M.; Ni, J.D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence—Its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Guo, M.L.; Buch, S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells 2019, 8, 1168. [Google Scholar] [CrossRef] [PubMed]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J. Virol. 2018, 92, e00993–18. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Au, K.Y.; Fang, J.W.; Yim, H.C.; Chow, K.H.; Ho, P.L.; Lau, A.S. HIV-1 trans-activator protein dysregulates IFN-gamma signaling and contributes to the suppression of autophagy induction. AIDS 2011, 25, 15–25. [Google Scholar] [CrossRef]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshetty, U.M.; Chatterjee, N.; Buch, S.; Periyasamy, P. HIV-1 Tat-Mediated Human Müller Glial Cell Senescence Involves Endoplasmic Reticulum Stress and Dysregulated Autophagy. Viruses 2024, 16, 903. https://doi.org/10.3390/v16060903
Deshetty UM, Chatterjee N, Buch S, Periyasamy P. HIV-1 Tat-Mediated Human Müller Glial Cell Senescence Involves Endoplasmic Reticulum Stress and Dysregulated Autophagy. Viruses. 2024; 16(6):903. https://doi.org/10.3390/v16060903
Chicago/Turabian StyleDeshetty, Uma Maheswari, Nivedita Chatterjee, Shilpa Buch, and Palsamy Periyasamy. 2024. "HIV-1 Tat-Mediated Human Müller Glial Cell Senescence Involves Endoplasmic Reticulum Stress and Dysregulated Autophagy" Viruses 16, no. 6: 903. https://doi.org/10.3390/v16060903
APA StyleDeshetty, U. M., Chatterjee, N., Buch, S., & Periyasamy, P. (2024). HIV-1 Tat-Mediated Human Müller Glial Cell Senescence Involves Endoplasmic Reticulum Stress and Dysregulated Autophagy. Viruses, 16(6), 903. https://doi.org/10.3390/v16060903