


Accurate Recapitulation of Chikungunya Virus Complete Coding Sequence Phylogeny Using Variable Genome Regions for Genomic Surveillance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Variability Analysis
2.2. Phylogenetic Analysis
2.3. Recapitulating the Phylogeny of the CDS
3. Results
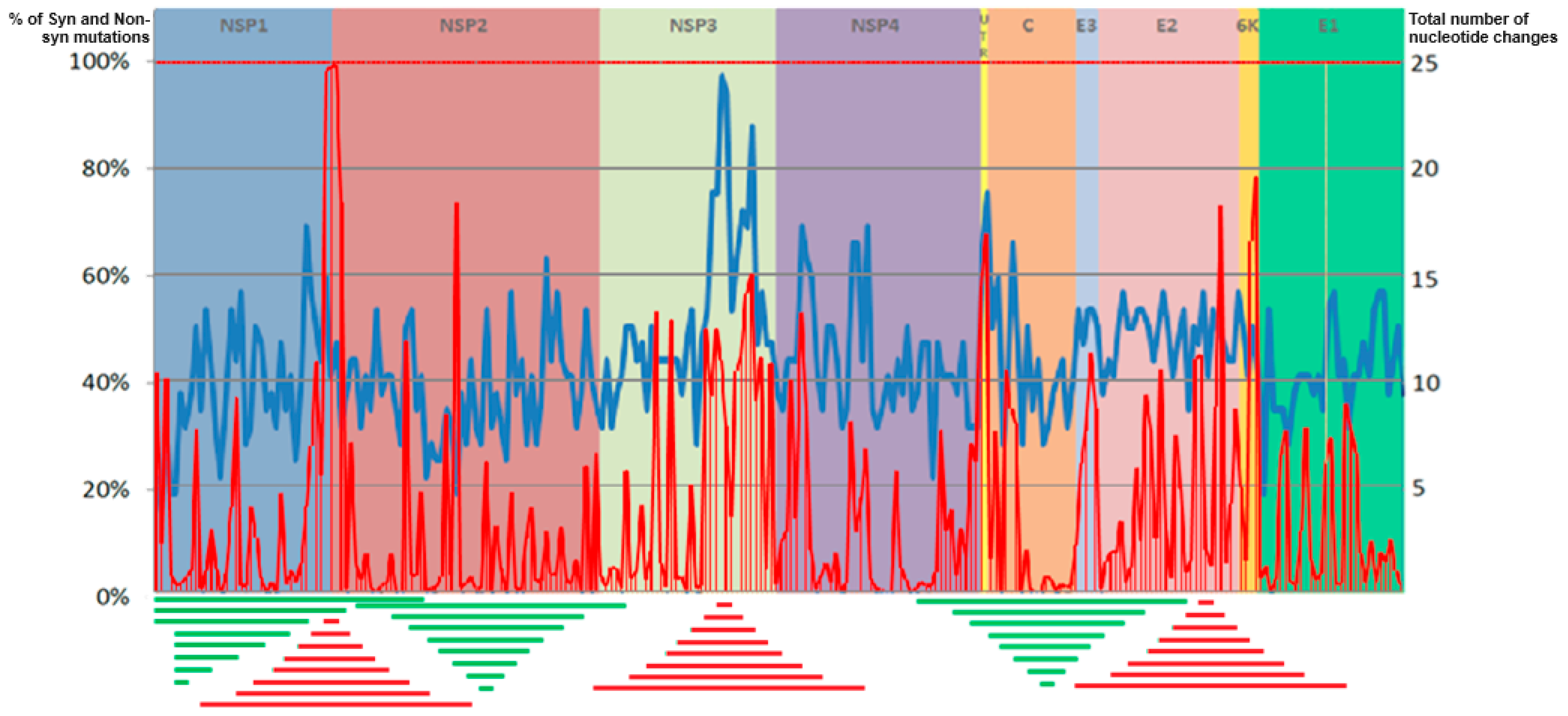
3.1. Genetic Variability Analysis
3.2. Evaluation of the Suitability of Hypervariable Regions for Recapitulating CDS Phylogeny
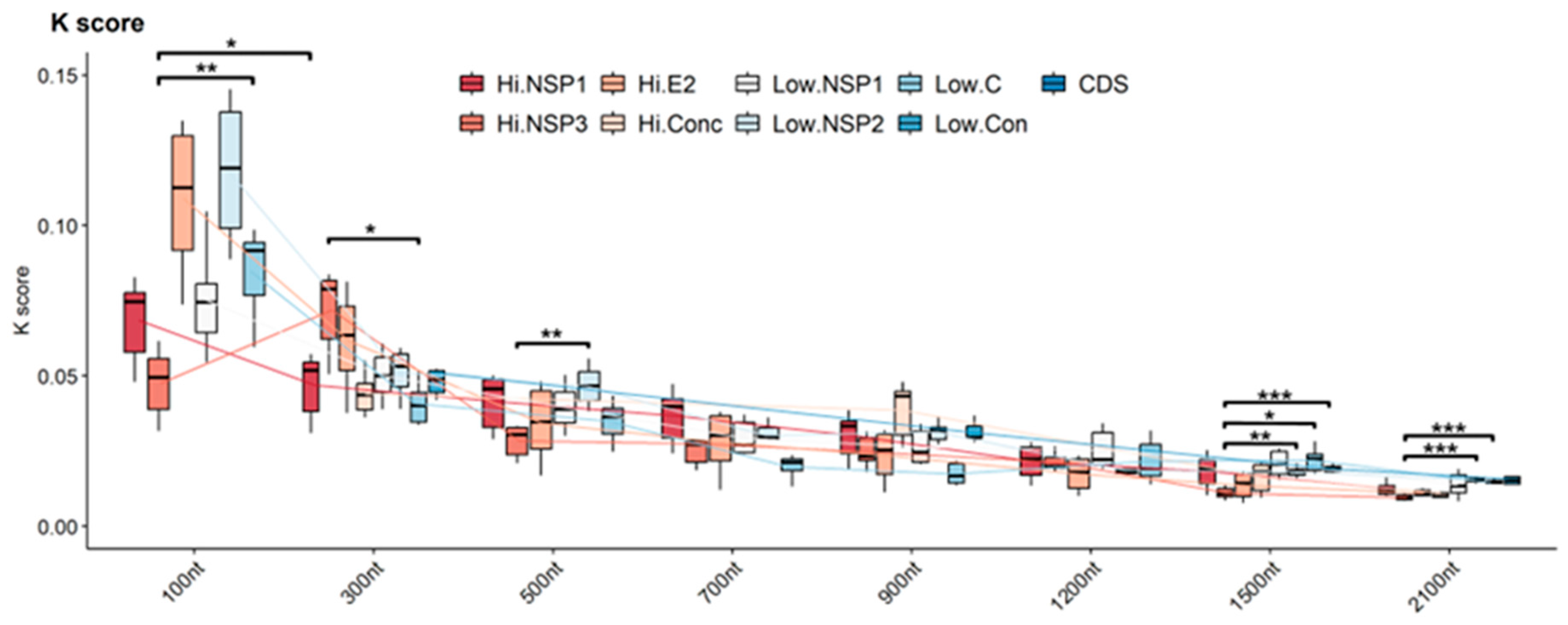
3.2.1. Branch Length Distance
3.2.2. Topological Incongruence
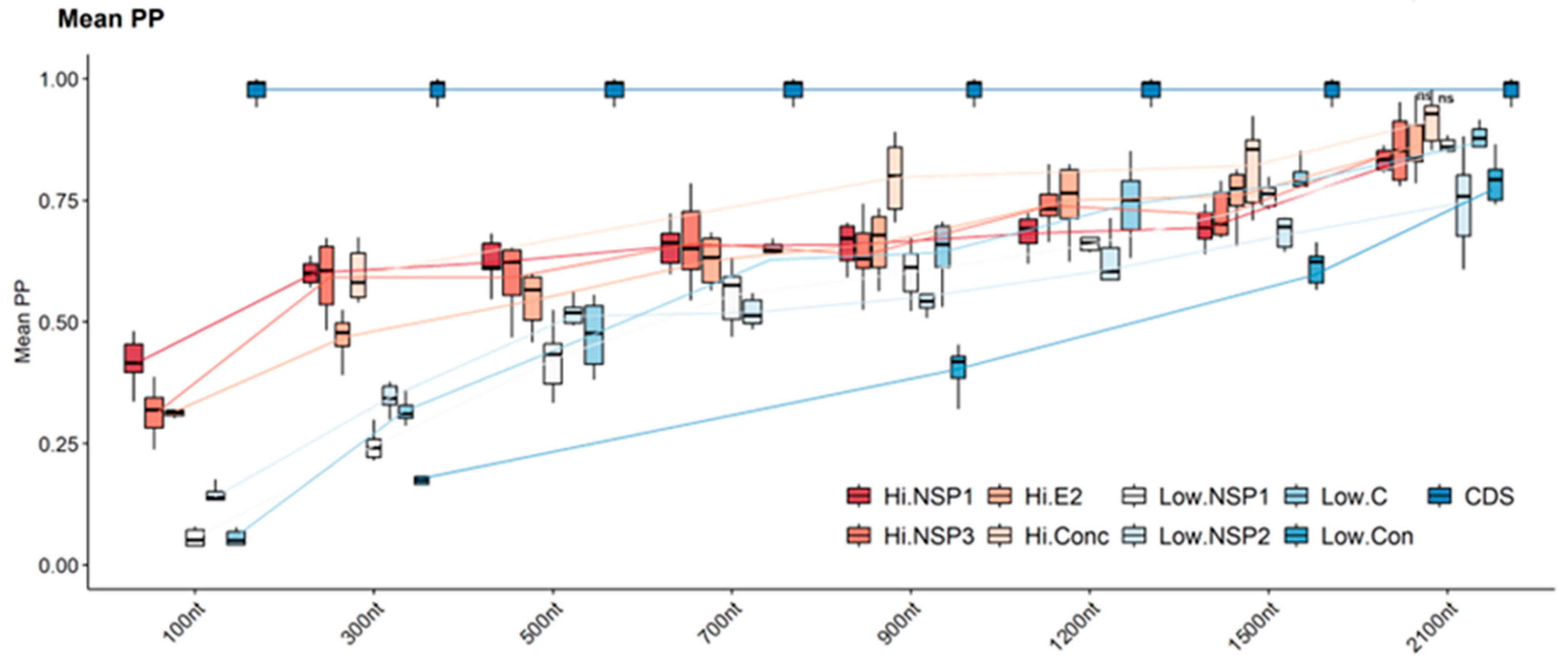
3.2.3. Tree Confidence
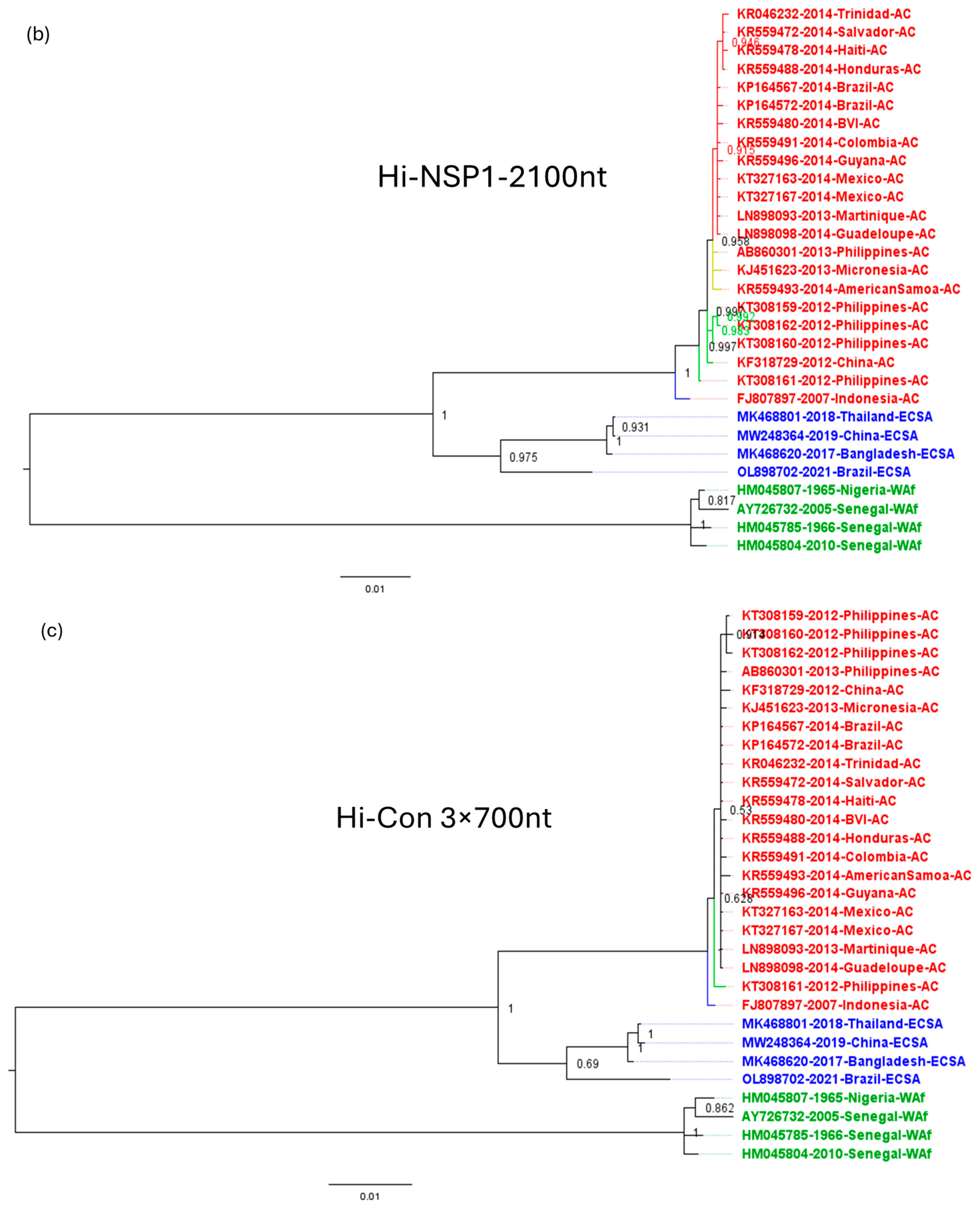
3.3. Inferring the Evolutionary Relationships among CHIKV Isolates from the American Outbreak Using Genomic Regions of Different Variability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chikungunya: Case Definitions for Acute, Atypical and Chronic Cases. Conclusions of an Expert Consultation, Managua, Nicaragua, 20–21 May 2015. Wkly. Epidemiol. Rec. 2015, 90, 410–414.
- Powers, A.M.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.; Strauss, J.H.; Weaver, S.C. Evolutionary Relationships and Systematics of the Alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef] [PubMed]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA Synthesis and Non-Structural Protein Functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Ross, R.W. The Newala Epidemic. III. The Virus: Isolation, Pathogenic Properties and Relationship to the Epidemic. J. Hyg. 1956, 54, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-Scale Phylogenetic Analyses of Chikungunya Virus Reveal Independent Emergences of Recent Epidemics and Various Evolutionary Rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Brault, A.C.; Tesh, R.B.; Weaver, S.C. Re-Emergence of Chikungunya and o’nyong-Nyong Viruses: Evidence for Distinct Geographical Lineages and Distant Evolutionary Relationships. J. Gen. Virol. 2000, 81, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Arankalle, V.A.; Shrivastava, S.; Cherian, S.; Gunjikar, R.S.; Walimbe, A.M.; Jadhav, S.M.; Sudeep, A.B.; Mishra, A.C. Genetic Divergence of Chikungunya Viruses in India (1963–2006) with Special Reference to the 2005–2006 Explosive Epidemic. J. Gen. Virol. 2007, 88, 1967–1976. [Google Scholar] [CrossRef]
- Rezza, G.; Nicoletti, L.; Angelini, R.; Romi, R.; Finarelli, A.C.; Panning, M.; Cordioli, P.; Fortuna, C.; Boros, S.; Magurano, F.; et al. Infection with Chikungunya Virus in Italy: An Outbreak in a Temperate Region. Lancet 2007, 370, 1840–1846. [Google Scholar] [CrossRef]
- Grandadam, M.; Caro, V.; Plumet, S.; Thiberge, J.-M.; Souarès, Y.; Failloux, A.-B.; Tolou, H.J.; Budelot, M.; Cosserat, D.; Leparc-Goffart, I.; et al. Chikungunya Virus, Southeastern France. Emerg. Infect. Dis. 2011, 17, 910–913. [Google Scholar] [CrossRef]
- Carey, D.E. Chikungunya and Dengue: A Case of Mistaken Identity? J. Hist. Med. Allied Sci. 1971, 26, 243–262. [Google Scholar] [CrossRef]
- Mehdi, Z.; Shahbaz, H.; Owais, A.; Hasan, S.U.; Nasr, I.; Jahangir, A.; Zubair, N.; Abdul Khaliq, S.A.; Khalid, M.; Shahbaz, S.; et al. Frequency, Awareness, and Symptoms of Chikungunya Among Patients in a Tertiary Care Hospital of Karachi: A Cross-Sectional Study. Cureus 2019, 11, e4054. [Google Scholar] [CrossRef] [PubMed]
- Leparc-Goffart, I.; Nougairede, A.; Cassadou, S.; Prat, C.; de Lamballerie, X. Chikungunya in the Americas. Lancet 2014, 383, 514. [Google Scholar] [CrossRef] [PubMed]
- Paixão, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, Chikungunya and Dengue: The Causes and Threats of New and Re-Emerging Arboviral Diseases. BMJ Glob. Health 2018, 3, e000530. [Google Scholar] [CrossRef] [PubMed]
- Yactayo, S.; Staples, J.E.; Millot, V.; Cibrelus, L.; Ramon-Pardo, P. Epidemiology of Chikungunya in the Americas. J. Infect. Dis. 2016, 214, S441–S445. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, M.; Moutailler, S.; Coudrier, D.; Rousseaux, C.; Khun, H.; Huerre, M.; Thiria, J.; Dehecq, J.-S.; Fontenille, D.; Schuffenecker, I.; et al. Two Chikungunya Isolates from the Outbreak of La Reunion (Indian Ocean) Exhibit Different Patterns of Infection in the Mosquito, Aedes Albopictus. PLoS ONE 2007, 2, e1168. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C. Arrival of Chikungunya Virus in the New World: Prospects for Spread and Impact on Public Health. PLoS Negl. Trop. Dis. 2014, 8, e2921. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Moratorio, G.; Henningsson, R.; Chen, R.; Matheus, S.; Enfissi, A.; Weissglas-Volkov, D.; Isakov, O.; Blanc, H.; Mounce, B.C.; et al. Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements. PLoS Negl. Trop. Dis. 2016, 10, e0004402. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Valadere, A.M. Transcontinental Movement of Asian Genotype Chikungunya Virus. Emerg. Infect. Dis. 2014, 20, 1400–1402. [Google Scholar] [CrossRef] [PubMed]
- Sahadeo, N.; Mohammed, H.; Allicock, O.M.; Auguste, A.J.; Widen, S.G.; Badal, K.; Pulchan, K.; Foster, J.E.; Weaver, S.C.; Carrington, C.V.F. Molecular Characterisation of Chikungunya Virus Infections in Trinidad and Comparison of Clinical and Laboratory Features with Dengue and Other Acute Febrile Cases. PLoS Negl. Trop. Dis. 2015, 9, e0004199. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Aguilar, E.D.; Martínez-Barnetche, J.; González-Bonilla, C.R.; Tellez-Sosa, J.M.; Argotte-Ramos, R.; Rodríguez, M.H. Genetic Diversity and Spatiotemporal Dynamics of Chikungunya Infections in Mexico during the Outbreak of 2014–2016. Viruses 2021, 14, 70. [Google Scholar] [CrossRef]
- Díaz, Y.; Carrera, J.-P.; Cerezo, L.; Arauz, D.; Guerra, I.; Cisneros, J.; Armién, B.; Botello, A.M.; Araúz, A.B.; Gonzalez, V.; et al. Chikungunya Virus Infection: First Detection of Imported and Autochthonous Cases in Panama. Am. J. Trop. Med. Hyg. 2015, 92, 482–485. [Google Scholar] [CrossRef]
- Mattar, S.; Miranda, J.; Pinzon, H.; Tique, V.; Bolanos, A.; Aponte, J.; Arrieta, G.; Gonzalez, M.; Barrios, K.; Contreras, H.; et al. Outbreak of Chikungunya Virus in the North Caribbean Area of Colombia: Clinical Presentation and Phylogenetic Analysis. J. Infect. Dev. Ctries. 2015, 9, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.T.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.G.; de Oliveira, L.F.; Azevedo, R.d.S.d.S.; da Silva, D.E.A.; da Silva, E.V.P.; da Silva, S.P.; et al. Emergence and Potential for Spread of Chikungunya Virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef]
- Langsjoen, R.M.; Haller, S.L.; Roy, C.J.; Vinet-Oliphant, H.; Bergren, N.A.; Erasmus, J.H.; Livengood, J.A.; Powell, T.D.; Weaver, S.C.; Rossi, S.L. Chikungunya Virus Strains Show Lineage-Specific Variations in Virulence and Cross-Protective Ability in Murine and Nonhuman Primate Models. mBio 2018, 9, e02449-17. [Google Scholar] [CrossRef]
- Sim, S.; Hibberd, M.L. Genomic Approaches for Understanding Dengue: Insights from the Virus, Vector, and Host. Genome Biol. 2016, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Holland, J.J. Mutation Rates among RNA Viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Robishaw, J.D.; Alter, S.M.; Solano, J.J.; Shih, R.D.; DeMets, D.L.; Maki, D.G.; Hennekens, C.H. Genomic Surveillance to Combat COVID-19: Challenges and Opportunities. Lancet Microbe 2021, 2, e481–e484. [Google Scholar] [CrossRef]
- Agarwal, A.; Sharma, A.K.; Sukumaran, D.; Parida, M.; Dash, P.K. Two Novel Epistatic Mutations (E1:K211E and E2:V264A) in Structural Proteins of Chikungunya Virus Enhance Fitness in Aedes Aegypti. Virology 2016, 497, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are Whole-Exome and Whole-Genome Sequencing Approaches Cost-Effective? A Systematic Review of the Literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Aguilar, E.D.; Martínez-Barnetche, J.; Rodríguez, M.H. Three Highly Variable Genome Regions of the Four Dengue Virus Serotypes Can Accurately Recapitulate the CDS Phylogeny. MethodsX 2022, 9, 101859. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the Model of DNA Substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Kuhner, M.K.; Felsenstein, J. A Simulation Comparison of Phylogeny Algorithms under Equal and Unequal Evolutionary Rates. Mol. Biol. Evol. 1994, 11, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Soria-Carrasco, V.; Talavera, G.; Igea, J.; Castresana, J. The K Tree Score: Quantification of Differences in the Relative Branch Length and Topology of Phylogenetic Trees. Bioinformatics 2007, 23, 2954–2956. [Google Scholar] [CrossRef]
- Alfaro, M.E.; Zoller, S.; Lutzoni, F. Bayes or Bootstrap? A Simulation Study Comparing the Performance of Bayesian Markov Chain Monte Carlo Sampling and Bootstrapping in Assessing Phylogenetic Confidence. Mol. Biol. Evol. 2003, 20, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.-C.; Lavenir, R.; Pardigon, N.; Reynes, J.-M.; Pettinelli, F.; et al. Genome Microevolution of Chikungunya Viruses Causing the Indian Ocean Outbreak. PLoS Med. 2006, 3, e263. [Google Scholar] [CrossRef]
- Torres-Longoria, B.; Fragoso-Fonseca, D.E.; Núñez-León, A.; de la Luz Torres, M.; Vázquez-Pichardo, M.; Escobar-Escamilla, N.; Wong-Arámbula, C.; Ramírez-González, J.E.; Méndez-Tenorio, A.; Castro-Mussot, M.E.; et al. Epidemiological Surveillance of Chikungunya Fever in Mexico since Its Introduction in 2014–2016 and Identification of Circulating Genotypes. Mol. Biol. Rep. 2021, 48, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Popovich, K.J.; Snitkin, E.S. Whole Genome Sequencing-Implications for Infection Prevention and Outbreak Investigations. Curr. Infect. Dis. Rep. 2017, 19, 15. [Google Scholar] [CrossRef]
- Mboowa, G.; Mwesigwa, S.; Kateete, D.; Wayengera, M.; Nasinghe, E.; Katagirya, E.; Katabazi, A.F.; Kigozi, E.; Kirimunda, S.; Kamulegeya, R.; et al. Whole-Genome Sequencing of SARS-CoV-2 in Uganda: Implementation of the Low-Cost ARTIC Protocol in Resource-Limited Settings. F1000Res 2021, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fu, A.; Hu, B.; Tong, Y.; Liu, R.; Liu, Z.; Gu, J.; Xiang, B.; Liu, J.; Jiang, W.; et al. Nanopore Targeted Sequencing for the Accurate and Comprehensive Detection of SARS-CoV-2 and Other Respiratory Viruses. Small 2020, 16, e2002169. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, H.; Giovanetti, M.; Xavier, J.; Adelino, T.E.R.; Fonseca, V.; de Jesus, J.G.; de Jesus, R.; Freitas, C.; Peterka, C.R.L.; Campelo de Albuquerque, C.F.; et al. Retrospective Genomic Surveillance of Chikungunya Transmission in Minas Gerais State, Southeast Brazil. Microbiol. Spectr. 2022, 10, e0128522. [Google Scholar] [CrossRef] [PubMed]
- Betancur-R, R.; Naylor, G.J.P.; Ortí, G. Conserved Genes, Sampling Error, and Phylogenomic Inference. Syst. Biol. 2014, 63, 257–262. [Google Scholar] [CrossRef]
- Doyle, V.P.; Young, R.E.; Naylor, G.J.P.; Brown, J.M. Can We Identify Genes with Increased Phylogenetic Reliability? Syst. Biol. 2015, 64, 824–837. [Google Scholar] [CrossRef]
- Yang, Z. On the Best Evolutionary Rate for Phylogenetic Analysis. Syst. Biol. 1998, 47, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Quiñonez, J.A.; Escobar-Escamilla, N.; Ortíz-Alcántara, J.; Vázquez-Pichardo, M.; de la Luz Torres-Rodríguez, M.; Nuñez-León, A.; Torres-Longoria, B.; López-Martínez, I.; Ruiz-Matus, C.; Kuri-Morales, P.; et al. Identification of Asian Genotype of Chikungunya Virus Isolated in Mexico. Virus Genes 2016, 52, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Laredo-Tiscareño, S.V.; Machain-Williams, C.; Rodríguez-Pérez, M.A.; Garza-Hernandez, J.A.; Doria-Cobos, G.L.; Cetina-Trejo, R.C.; Bacab-Cab, L.A.; Tangudu, C.S.; Charles, J.; De Luna-Santillana, E.J.; et al. Arbovirus Surveillance near the Mexico–U.S. Border: Isolation and Sequence Analysis of Chikungunya Virus from Patients with Dengue-like Symptoms in Reynosa, Tamaulipas. Am. J. Trop. Med. Hyg. 2018, 99, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-Scale Approaches to Resolving Incongruence in Molecular Phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Medina, J.E.; Garcia-Knight, M.A.; Sanchez-Flores, A.; Monroy-Muñoz, I.E.; Grande, R.; Esbjörnsson, J.; Santacruz-Tinoco, C.E.; González-Bonilla, C.R. Evolutionary Analysis of the Chikungunya Virus Epidemic in Mexico Reveals Intra-Host Mutational Hotspots in the E1 Protein. PLoS ONE 2018, 13, e0209292. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, C.; Qiu, Y.; Liao, R.; Xuan, Z.; Ren, F.; Dong, Y.; Xie, X.; Han, Y.; Wu, D.; et al. Conserved Untranslated Regions of Multipartite Viruses: Natural Markers of Novel Viral Genomic Components and Tags of Viral Evolution. Virus Evol. 2024, 10, veae004. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, E.; Tsetsarkin, K.A.; Weaver, S.C. Chikungunya Virus 3’ Untranslated Region: Adaptation to Mosquitoes and a Population Bottleneck as Major Evolutionary Forces. PLoS Pathog. 2013, 9, e1003591. [Google Scholar] [CrossRef]
- Duchêne, D.A.; Mather, N.; Van Der Wal, C.; Ho, S.Y.W. Excluding Loci with Substitution Saturation Improves Inferences from Phylogenomic Data. Syst. Biol. 2022, 71, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef] [PubMed]
- Binet, M.; Gascuel, O.; Scornavacca, C.; Douzery, E.J.P.; Pardi, F. Fast and Accurate Branch Lengths Estimation for Phylogenomic Trees. BMC Bioinform. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Rodpothong, P.; Auewarakul, P. Positive Selection Sites in the Surface Genes of Dengue Virus: Phylogenetic Analysis of the Interserotypic Branches of the Four Serotypes. Virus Genes 2012, 44, 408–414. [Google Scholar] [CrossRef]
- Mindell, D.P.; Thacker, C.E. Rates of Molecular Evolution: Phylogenetic Issues and Applications. Annu. Rev. Ecol. Syst. 1996, 27, 279–303. [Google Scholar] [CrossRef]
- Som, A. Causes, Consequences and Solutions of Phylogenetic Incongruence. Brief. Bioinform. 2015, 16, 536–548. [Google Scholar] [CrossRef]
- Gontcharov, A.A.; Marin, B.; Melkonian, M. Are Combined Analyses Better than Single Gene Phylogenies? A Case Study Using SSU rDNA and rbcL Sequence Comparisons in the Zygnematophyceae (Streptophyta). Mol. Biol. Evol. 2004, 21, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Meredith, L.W.; Aboualy, M.; Ochola, R.; Okwarah, P.; Ozel, M.; Abubakar, A.; Barakat, A. Key Aspects Defining the Development and Implementation of a Regional Genomic Surveillance Strategy for the Eastern Mediterranean Region. Influenza Other Respir. Viruses 2023, 17, e13205. [Google Scholar] [CrossRef] [PubMed]
- Seib, K.L.; Dougan, G.; Rappuoli, R. The Key Role of Genomics in Modern Vaccine and Drug Design for Emerging Infectious Diseases. PLoS Genet. 2009, 5, e1000612. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.L.; MacCannell, D.R.; Carleton, H.A.; Neuhaus, E.B.; Bradbury, R.S.; Posey, J.E.; Taylor, J.; Gwinn, M. Pathogen Genomics in Public Health. N. Engl. J. Med. 2019, 381, 2569–2580. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Aguilar, E.D.; Gutiérrez-Millán, E.; Rodríguez, M.H. Accurate Recapitulation of Chikungunya Virus Complete Coding Sequence Phylogeny Using Variable Genome Regions for Genomic Surveillance. Viruses 2024, 16, 926. https://doi.org/10.3390/v16060926
Rodríguez-Aguilar ED, Gutiérrez-Millán E, Rodríguez MH. Accurate Recapitulation of Chikungunya Virus Complete Coding Sequence Phylogeny Using Variable Genome Regions for Genomic Surveillance. Viruses. 2024; 16(6):926. https://doi.org/10.3390/v16060926
Chicago/Turabian StyleRodríguez-Aguilar, Eduardo D., Everardo Gutiérrez-Millán, and Mario H. Rodríguez. 2024. "Accurate Recapitulation of Chikungunya Virus Complete Coding Sequence Phylogeny Using Variable Genome Regions for Genomic Surveillance" Viruses 16, no. 6: 926. https://doi.org/10.3390/v16060926
APA StyleRodríguez-Aguilar, E. D., Gutiérrez-Millán, E., & Rodríguez, M. H. (2024). Accurate Recapitulation of Chikungunya Virus Complete Coding Sequence Phylogeny Using Variable Genome Regions for Genomic Surveillance. Viruses, 16(6), 926. https://doi.org/10.3390/v16060926